本文来自微信公众号:X-MOLNews
手性 N -烷基吲哚骨架广泛存在于天然产物和药物分子中,同时还是重要的有机中间体。然而,从简单易得的原料和催化剂出发选择性合成手性 N -烷基吲哚分子长期以来是有机合成和药物研发领域的一大难题。由于芳香共轭作用,吲哚N上亲核性降低,传统C-N偶联方法构建手性 N -烷基吲哚通常存在吲哚N1位和C3位的选择性竞争,因此,往往需要在吲哚上引入拉电子基团、C3位堵上取代基,或者通过间接方法合成。另一方面,虽然已有C-N偶联方法可以不同程度实现手性 N -苄基吲哚、 N -烯丙基吲哚、 N -炔丙基吲哚的不对称催化合成,但通常不同类型的手性 N -烷基吲哚合成需要不同的方法。鉴于此, 云南大学钱德云 (点击查看介绍)课题组基于C-C偶联和模块化合成策略,报道了一种 Ni-H催化的不对称氢碳*能官**团化反应:从简单易得的 N -烯基吲哚和芳基、烯基、炔基溴化物出发,原位形成 N -烷基镍中间体,实现形式上的C(sp3)–C(sp2)/C(sp)偶联,从而快速、高效、高选择性合成得到广泛的手性 N -烷基吲哚分子 。该反应条件温和,*能官**团兼容性广泛,能够对一系列天然产物、药物分子进行后期*能官**团化修饰,同时也能够用于快速合成生物活性分子类似物,如血清素再摄取*制剂抑**类似物。该工作近期发表在 Nat. Commun. 上。
在先前的报道中,虽然Melchiorre、Davidson课题组分别基于不同的 α-N -烷基自由基前体,通过Ni/光氧化还原共催化的C-C偶联可以实现手性 N -乙酰基吲哚、 N -苄基吲哚的合成,但底物范围较为局限,对映选择性普遍不高(42-82% ee)。在前期手性二烷基胺的不对称催化合成( J. Am. Chem. Soc . 2021 , 143 , 1959; ACS Catal . 2021 , 11 , 6560)工作基础上,作者发现利用简单易得的 N -烯基吲哚化合物在Ni-H催化下可以原位形成 α-N- 烷基镍物种,从而实现后续C-C偶联,通过催化剂与配体的调控化学、区域、立体选择性地构建手性 N -苄基、 N -烯丙基、 N -炔丙基吲哚分子骨架(最高达91% 产率和97% ee)。需要指出的是, N -烯基吲哚在以往的反应中多用于聚合反应合成高分子材料,也有可能双键被还原,从而使其参与的高效、高立体选择性不对称催化研究面临巨大挑战。
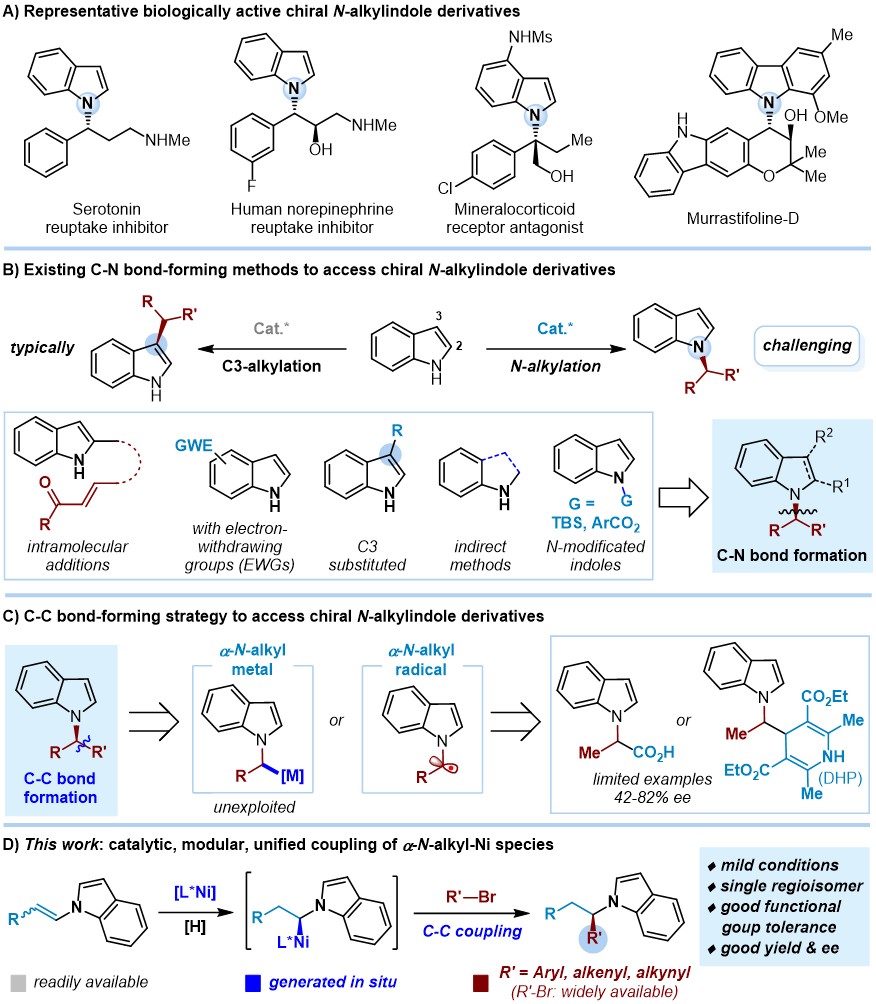
图1. 研究背景及该文工作
通过对不同的手性配体和其它反应参数进行筛选,与芳基/烯基溴化物反应,作者确定了最优的反应条件:以 N -烯基吲哚( 1 )、2.0倍当量溴化物( 2 )作为反应物,10 mol% NiI2·xH2O作为镍催化剂,15 mol%双噁唑啉Bi-Ox( L*1 )作为手性配体,加入1.2/ 1.5倍量 (OEt)2SiMeH/ KF,室温下在乙二醇二甲醚/二氯乙烷(DME/DCE,3/1)溶剂中进行反应,以19-79%的收率和58-97%对映选择性得到目标产物(图2)。这一方法具有较好的通用性,多样化的 N -烯基吲哚和芳基溴化物可以很好地兼容;无论是缺电子还是富电子的芳烃都可以用于偶联,其中取代基可以在对位、间位和邻位。各种*能官**团,如腈基( 2b )、三氟甲基( 2c、2p、2r )、酯( 2d、2m、2q )、卤化物( 2f、2g、2n、2o )、醚( 2i、2j ),特别是容易被还原的酮( 2a )、醛( 2e )以及易于发生偶联反应的三氟甲磺酸酯( 2k )、硼酸酯( 2l )等敏感*能官**团,在标准反应条件下都兼容。此外,具有重要药用意义的吡啶杂环( 2r )也适用。
接下来,作者研究了不同 N -烯基吲哚对反应的影响。在吲哚苯环的不同位置有不同的*能官**团,包括6-甲氧基( 1s )、6-氟( 1t )、6-氰基( 1u )、5-甲基( 1v )取代基以及烯基咔唑( 1x )都可以很好地兼容。 N -烯基吲哚的C3位被取代后,得到相应的产物 3w ,反应同样具有高的对映选择性,这在之前的Cu-H催化下很难得到。同时,二取代( Z )- N -烯基吲哚也能够被成功地转化,以中等的产率和高的对映选择性得到相应的 N -烷基吲哚( 3y-3dd )。( E )- N -烯基吲哚与( Z )- N -烯基吲哚相比,反应活性较低,对映选择性略低(例如, 3y :19%,84% ee vs . 41%,87% ee)。在反应过程中,可以观察到 Z 式烯基底物异构化为 E 式,反应后可以回收出40%以上的 E/Z 混合物的烯基吲哚。值得注意的是,即使烯基上存在其他导向基团,如酰胺( 3z-3bb )、酯( 3cc )以及芳基( 3dd ),C-C键的形成都会区域选择性地发生在 α -碳上。该模块化反应可用于制备重要的5-羟色胺再摄取*制剂抑**衍生物(如图4),具有良好的效率和对映选择性( 3z-3bb ,81-92%ee)。
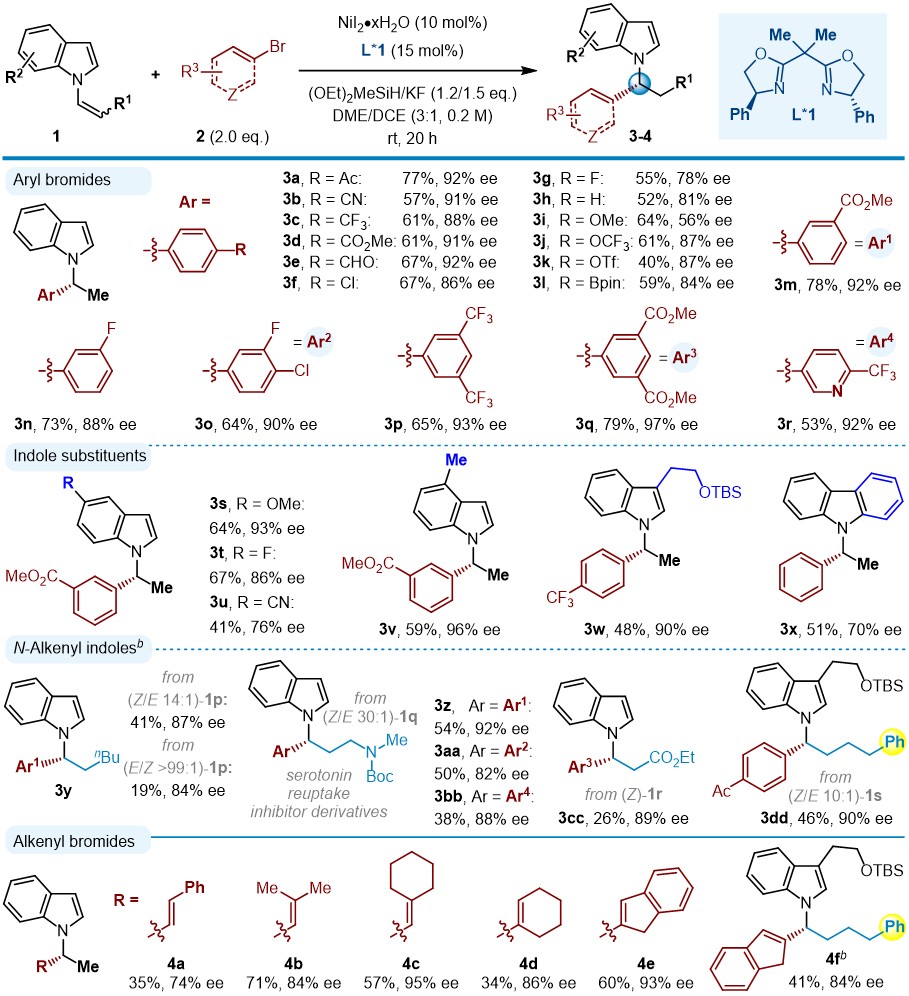
图2. 手性 N -苄基吲哚和 N -烯丙基吲哚的合成
除了芳基溴化物外,这一催化C-C偶联反应也适用于烯基溴 4 和炔基溴 5 ,使手性 N -烷基吲哚结构多样化。特别是,这反应补充了之前建立的金属催化的吲哚 N -烯丙化的方法,使得含有芳基和烷基的二取代和三取代烯丙基产物很容易获得。此外,使用 L*8 配体,除了可以调控 N -炔丙基的合成以外,也可以高对映选择性地得到 N -苄基和 N -烯丙基吲哚产物(图3)。
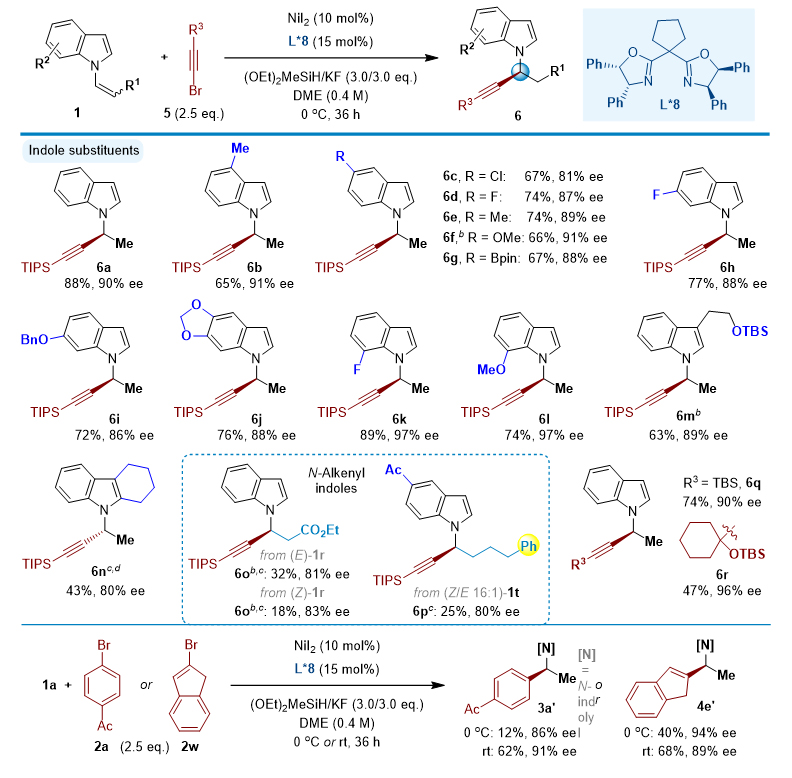
图3. 手性 N -炔丙基吲哚的合成
作者应用该方法还对多种天然产物和药物分子进行了后期多样化衍生以及*能官**团化(图4),制备了一系列源于生物活性分子的手性 N -烷基吲哚化合物( 7a-7e )。而且,可以进行克级规模合成。
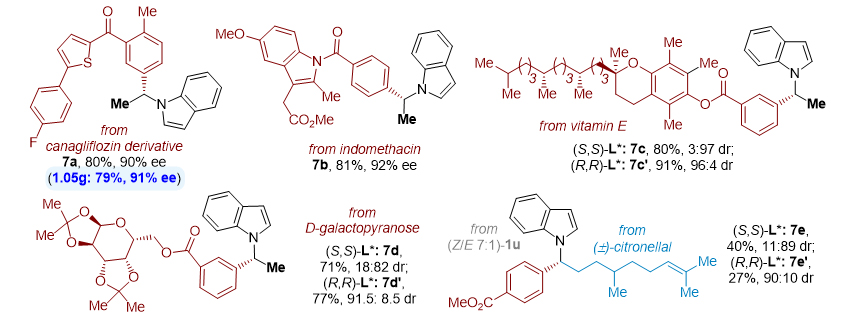
图4. 应用于天然产物和药物分子的修饰
此外,作者对 N -炔丙基吲哚进行了转化,通过去硅基保护剂、氢化、叠氮环化、Sonogashira-偶联分别得到了化合物 8-12 (如图5)。
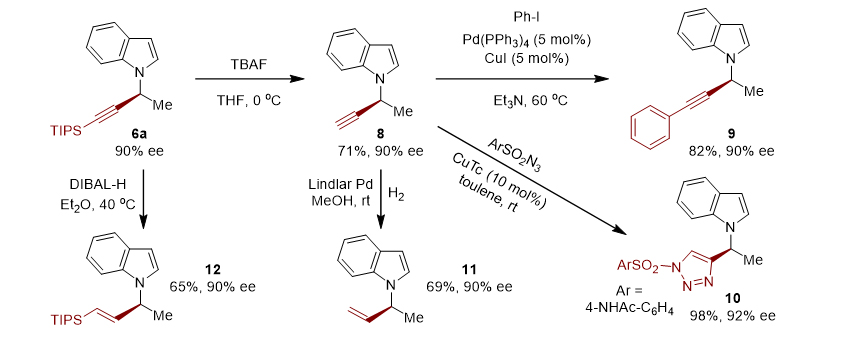
图5. 手性 N -炔丙基吲哚转化
为了进一步研究该反应的机理,作者进行了一系列的对照实验(图6a)、动力学研究(图6b)以及Hammett实验(图6c)。实验表明:芳基亲电试剂的对位取代基变化对反应速率有显著影响,反应的氧化加成很有可能是速率决速步骤。
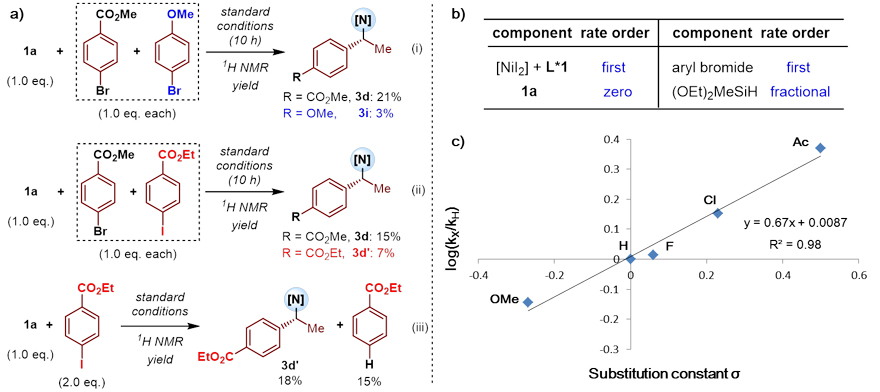
图6. 对照实验和Hammett实验
非线性效应研究(图7a)表明:单一催化剂是真正的活性物种。不同芳基溴化物的Hammett取代基参数与相应产物的对映体过量之间的线性关系(图7b)进一步表明:氧化加成还有可能是反应的对映选择性决定步骤。据此,作者提出了如图8所示的可能机理。
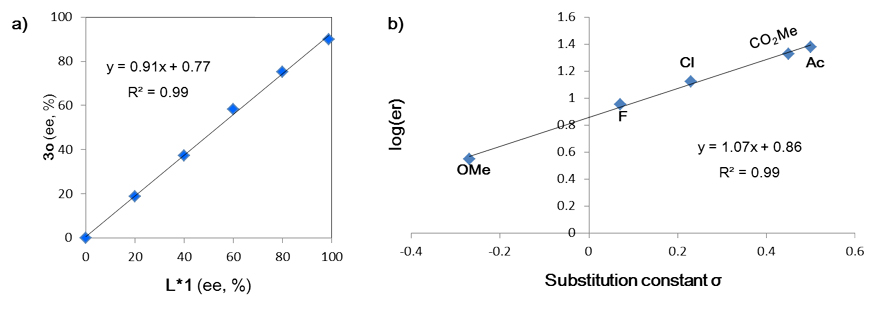
图7. 非线性效应和Hammett线性关系

图8. 可能的反应机理
总结
钱德云课题组通过镍催化烯基吲哚与芳基、烯基、炔基溴化物偶联构建C-C键,发展了一种对映选择性、模块化合成手性 N -烷基吲哚的新策略与新方法。该反应在温和条件下经由原位生成 α-N -烷基镍物种而实现,具有较好的*能官**团耐受性和底物普适性。复杂药物分子和天然产物的后期多样化衍生以及*能官**团化,进一步证明了这一方法在合成有价值的手性 N -烷基化生物活性分子中的*在用潜**途。这一工作是与 邵志会 课题组、 羊晓东 课题组合作共同完成的,硕士研究生 李伦、任江涛 为论文共同第一作者。
Enantioselective synthesis of N -alkylindoles enabled by nickel-catalyzed C-C coupling
Lun Li, Jiangtao Ren, Jingjie Zhou, Xiaomei Wu, Zhihui Shao, Xiaodong Yang & Deyun Qian
Nat. Commun ., 2022 , 13 , 6861. DOI: 10.1038/s41467-022-34615-9
导师介绍
钱德云
http://www.lmcnr.ynu.edu.cn/info/1006/2043.htm
https://www.x-mol.com/groups/Qian_Deyun