说明:以二维InSe原胞为例,QE版本:qe-6.4.1,赝势:SSSP。
1. 结构优化
通过Materials studio 2018的建模功能切出二维InSe原胞,如图所示,并导出为InSe.cif格式。

二维InSe原胞
使用VESTA软件打开InSe.cif文件,然后File→Export Date,保存格式选择vasp,原子坐标选择Fractional coordinates,最后点击确定。
2、QE输入文件结构
QE的结构优化主要使用的是pw.x模块,该模块包含非常多的输入参数,建议在计算过程中逐步学习,无需一次性学完所有命令,即使学完也是徒劳,因为几乎无法记住。
结构优化在QE中是比较诟病的一个地方,优化速度非常慢,对于大体系实现结构优化是非常慢的,如果计算的体系包含较多的原子,建议使用VASP。QE结合EPW计算材料的与声子有关的性质比较合适,且这类计算一般不需要建立超胞,能够达到计算目的。总之,根据计算的任务和目的合理选择第一性原理计算软件是比较明智的。
2.1 pw.x模块输入文件结构
&CONTROL.../&SYSTEM.../&ELECTRONS.../[ &IONS.../ ][ &CELL.../ ]ATOMIC_SPECIESX Mass_X PseudoPot_XY Mass_Y PseudoPot_YZ Mass_Z PseudoPot_ZATOMIC_POSITIONS { alat | bohr | crystal | angstrom | crystal_sg }X 0.0 0.0 0.0 {if_pos(1) if_pos(2) if_pos(3)}Y 0.5 0.0 0.0Z O.0 0.2 0.2K_POINTS { tpiba | automatic | crystal | gamma | tpiba_b | crystal_b | tpiba_c | crystal_c }if (gamma)nothing to readif (automatic)nk1, nk2, nk3, k1, k2, k3if (not automatic)nksxk_x, xk_y, xk_z, wk[ CELL_PARAMETERS { alat | bohr | angstrom }v1(1) v1(2) v1(3)v2(1) v2(2) v2(3)v3(1) v3(2) v3(3) ][ OCCUPATIONSf_inp1(1) f_inp1(2) f_inp1(3) ... f_inp1(10)f_inp1(11) f_inp1(12) ... f_inp1(nbnd)[ f_inp2(1) f_inp2(2) f_inp2(3) ... f_inp2(10)f_inp2(11) f_inp2(12) ... f_inp2(nbnd) ] ][ CONSTRAINTSnconstr { constr_tol }constr_type(.) constr(1,.) constr(2,.) [ constr(3,.) constr(4,.) ] { constr_target(.) } ][ ATOMIC_FORCESlabel_1 Fx(1) Fy(1) Fz(1).....label_n Fx(n) Fy(n) Fz(n) ]
2.2 二维InSe原胞结构优化输入文件
&CONTROLcalculation='vc-relax', ! vc-relax | relaxrestart_mode='from_scratch', ! normal usedprefix='InSe', ! prepended to input/output filenamespseudo_dir='../pseudo/', ! directory containing pseudopotential filesoutdir='../tmp/', ! input, temporary, output files are found in this directoryforc_conv_thr=1d-5, ! forces convergence threshold 1d-03/&SYSTEMibrav = 0 ! Bravais-lattice indexnat=4 ! number of atoms in the unit cellntyp=2 ! number of types of atoms in the unit cellecutwfc=60.0, ! kinetic energy cutoff (Ry) for wavefunctionsecutrho=720.0, ! Kinetic energy cutoff (Ry) for charge density/&ELECTRONSmixing_beta=0.7, ! mixing factor for self-consistencyconv_thr=1d-8, ! Convergence threshold for selfconsistency 1d-6/&IONSion_dynamics='bfgs', ! Specify the type of ionic dynamics/&CELLcell_dynamics='bfgs', ! Specify the type of dynamics for the cellpress=0.0, !press_conv_thr=0.5, ! Convergence threshold on the pressure for variable cellcell_dofree=2Dxy, ! for 2D materials/CELL_PARAMETERS {angstrom} ! specify the structure4.0836000443 0.0000000000 0.0000000000-2.0418000221 3.5365013772 0.00000000000.0000000000 0.0000000000 25.3775005341ATOMIC_SPECIES !In 114.80 In.pbe-dn-rrkjus_psl.0.2.2.UPFSe 78.960 Se_pbe_v1.uspp.F.UPFATOMIC_POSITIONS {crystal}In 0.333333347 0.666666695 0.555589958In 0.333333347 0.666666695 0.444410042Se 0.666666674 0.333333347 0.394049989Se 0.666666674 0.333333347 0.605950011K_POINTS automatic12 12 1 0 0 0
2.3 结构优化的经验总结
pw.x模块用于结构优化时,对于体系的对称性非常敏感,也就是QE自身不会寻找体系的对称性,只能依靠手动输入体系的对称性,这点与VASP相比是非常欠缺的,因为VASP是可以自己寻找对称性的。对于二维InSe原胞,有对称性与无对称性的结构优化使用时间相差了近5倍。
查看优化所用时间
grep PWSCF InSe.vc-relax.out | tail -n 1
输入对称性
PWSCF : 4m10.53s CPU 4m51.82s WALL
没有输入对称性
PWSCF : 21m5.53s CPU 4m51.82s WALL
对于离子的受力,一般使用准牛顿迭代算法
ion_dynamics='bfgs'
pwscf模块做结构优化时,对于二维材料无需重新编译软件,即可做到不优化真空层,这点比VASP方便,具体设置如下
cell_dofree CHARACTERDefault: 'all'Select which of the cell parameters should be moved:'all' :all axis and angles are moved'ibrav' :all axis and angles are moved, but the lattice but be representable with the initial ibrav choice'x' :only the x component of axis 1 (v1_x) is moved'y' :only the y component of axis 2 (v2_y) is moved'z' :only the z component of axis 3 (v3_z) is moved'xy' :only v1_x and v2_y are moved'xz' :only v1_x and v3_z are moved'yz' :only v2_y and v3_z are moved'xyz' :only v1_x, v2_y, v3_z are moved'shape' :all axis and angles, keeping the volume fixed'volume' :the volume changes, keeping all angles fixed (i.e. only celldm(1) changes)'2Dxy' :only x and y components are allowed to change'2Dshape' :as above, keeping the area in xy plane fixed'epitaxial_ab' :fix axis 1 and 2 while allowing axis 3 to move'epitaxial_ac' :fix axis 1 and 3 while allowing axis 2 to move'epitaxial_bc' :fix axis 2 and 3 while allowing axis 1 to moveBEWARE: if axis are not orthogonal, some of these options do not work symmetryisbroken. If you are not happy with them, edit subroutine init_dofree in file Modules/cell_base.f90
3、结构自洽计算
3.1 输入文件
&CONTROLcalculation='scf',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../tmp/',/&SYSTEMibrav = 0nat=4ntyp=2ecutwfc=60.0,ecutrho=480.0,/&ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS {angstrom}4.081910567 0.000000000 0.000000000-2.040955283 3.535038247 0.0000000000.000000000 0.000000000 25.377500534ATOMIC_SPECIESIn 114.80 In.pbe-dn-rrkjus_psl.0.2.2.UPFSe 78.960 Se_pbe_v1.uspp.F.UPFATOMIC_POSITIONS {crystal}In 0.333333347 0.666666695 0.555619036In 0.333333347 0.666666695 0.444380964Se 0.666666674 0.333333347 0.394016400Se 0.666666674 0.333333347 0.605983600K_POINTS automatic12 12 1 0 0 0mpirun -np 16 pw.x < InSe.scf.in.$ecutwfc > InSe.scf.out.$ecutwfcgrep -e 'kinetic-energy cutoff' -e ! InSe.scf.out.$ecutwfc | \awk '/kinetic/{ecutwfc=$(NF-1)}/!/{print ecutwfc, $(NF-1)}' >> InSe.etot_vs_ecutwfcdone
3.2 ecutwfc测试
for ecutwfc in 50 55 60 65 70 75 80 ; do# self-consistent calculationcat > InSe.scf.in.$ecutwfc << EOF&CONTROLcalculation='scf',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../test_tmp/',/&SYSTEMibrav = 0nat=4ntyp=2ecutwfc=$ecutwfc,ecutrho=$((8*$ecutwfc)),/&ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS {angstrom}4.081910567 0.000000000 0.000000000-2.040955283 3.535038247 0.0000000000.000000000 0.000000000 25.377500534ATOMIC_SPECIESIn 114.80 In.pbe-dn-rrkjus_psl.0.2.2.UPFSe 78.960 Se_pbe_v1.uspp.F.UPFATOMIC_POSITIONS {crystal}In 0.333333347 0.666666695 0.555619036In 0.333333347 0.666666695 0.444380964Se 0.666666674 0.333333347 0.394016400Se 0.666666674 0.333333347 0.605983600K_POINTS automatic12 12 1 0 0 0EOF
测试结果:
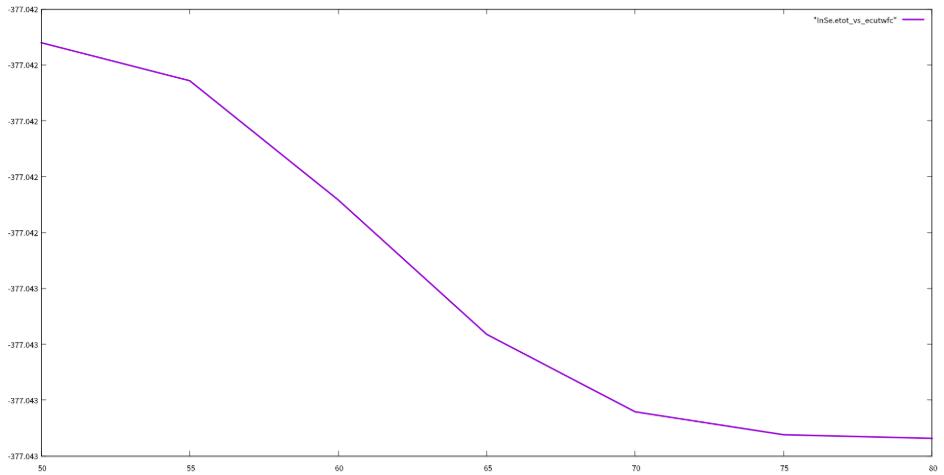
encut
3.3 kpoints测试
for kpoints in 4 6 8 10 12 14 16 18 20 ; do# self-consistent calculationcat > InSe.scf.in.$kpoints << EOF&CONTROLcalculation='scf',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../test_tmp/',/&SYSTEMibrav = 0nat=4ntyp=2ecutwfc=70,ecutrho=840,/&ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS {angstrom}4.081910567 0.000000000 0.000000000-2.040955283 3.535038247 0.0000000000.000000000 0.000000000 25.377500534ATOMIC_SPECIESIn 114.80 In.pbe-dn-rrkjus_psl.0.2.2.UPFSe 78.960 Se_pbe_v1.uspp.F.UPFATOMIC_POSITIONS {crystal}In 0.333333347 0.666666695 0.555619036In 0.333333347 0.666666695 0.444380964Se 0.666666674 0.333333347 0.394016400Se 0.666666674 0.333333347 0.605983600K_POINTS automatic$kpoints $kpoints 1 0 0 0EOFmpirun -np 16 pw.x < InSe.scf.in.$kpoints > InSe.scf.out.$kpointsecho -e "$kpoints \c" >> InSe.etot_vs_kpointsgrep ! InSe.scf.out.$kpoints | \awk '/!/{print $(NF-1)}' >> InSe.etot_vs_kpointsdone
测试结果:

kpoints
3.4 结构自洽计算总结
通过对ecutwfc和kpoints的测试,我们选定ecutwfc的值为80 Ry,kpoints的值为8 8 1,用这个值返回去重新优化结构和自洽,然后开始计算下面的性质。
4、能带计算
4.1 输入文件
&CONTROLcalculation='bands',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../tmp/',verbosity='high',/&SYSTEMibrav = 0nat=4ntyp=2ecutwfc=80.0,ecutrho=960.0,nbnd=26,/&ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS {angstrom}4.085963663 -0.000000000 0.000000000-2.042981832 3.538548331 0.0000000000.000000000 0.000000000 25.377500534ATOMIC_SPECIESIn 114.80 In.pbe-dn-rrkjus_psl.0.2.2.UPFSe 78.960 Se_pbe_v1.uspp.F.UPFATOMIC_POSITIONS {crystal}In 0.333333347 0.666666695 0.555623200In 0.333333347 0.666666695 0.444376800Se 0.666666674 0.333333347 0.394099781Se 0.666666674 0.333333347 0.605900219K_POINTS crystal_b40.5 0.0 0.0 100 !M0.0 0.0 0.0 100 !G0.33333 0.33333 0.0 100 !K0.5 0.0 0.0 100 !M
4.2 后处理文件band2.in文件
&bandsprefix='InSe',outdir='../tmp/',filband='InSebands.dat',lsym=.true./
4.3 画能带图的后处理程序plotband.x
> plotband.x InSebands.datReading 26 bands at 301 k-pointsfile with representations not compatible with bandsRange: -16.4500 5.4820eV Emin, Emax > -4 4high-symmetry point: 0.5000 0.2887 0.0000 x coordinate 0.0000high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.5774high-symmetry point: 0.3333 0.5773 0.0000 x coordinate 1.2440high-symmetry point: 0.5000 0.2887 0.0000 x coordinate 1.5773output file (gnuplot/xmgr) > InSebands.plotbands in gnuplot/xmgr format written to file InSebands.plotoutput file (ps) > InSebands.psEfermi > -1.5444deltaE, reference E (for tics) 2 -1.5444bands in PostScript format written to file InSebands.ps
说明:QE自带的画图程序plotband.x画出的能带图几乎无法直接用于文章中,因此这步处理意义不大。可以直接使用band.x计算出的InSebands.dat.gnu文件,通过origin等专业的绘图软件绘制能带图。
能带图计算结果:
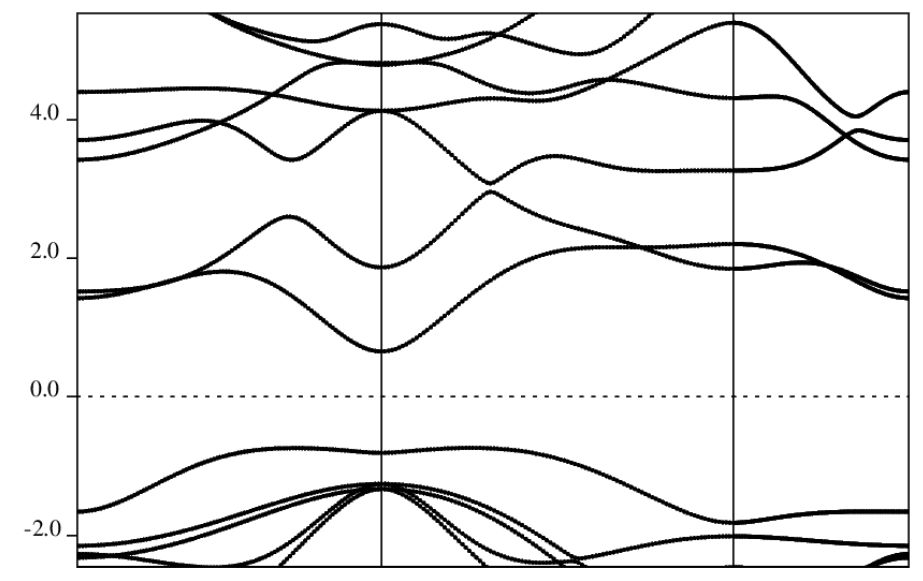
2D_InSe-band_qe
本文章转载自贺勇个人博客,请大家多多关注,有非常多的干货!

链接: https://yh-phys.github.io/2019/10/11/qe-band/
5、其它QE入门教程友情链接
(1) https://yyyu200.github.io/DFTbook/
(2) http://blog.sciencenet.cn/u/pfliu89
(3) http://blog.sciencenet.cn/u/yhli0906