来源:中华儿科杂志
摘要
目的
总结先天性肺囊性疾病的诊断、临床表现、治疗及预后。
方法
回顾分析在天津市儿童医院2010年1月至2019年3月住院治疗的96例先天性肺囊性疾病患儿(男50例、女46例)的临床表现、影像学检查、病理、治疗及预后。
结果
96例先天性肺囊性疾病患儿年龄4 d~13岁,86例(90%)患儿因咳嗽发热就诊时发现。先天性囊性腺瘤样畸形40例(42%),手术治疗30例,急症手术2例,死亡1例。肺隔离症12例(13%),手术治疗4例。先天性肺囊肿12例(13%),手术治疗4例。先天性大叶性肺气肿3例(3%),手术治疗1例,1例气胸患儿出院后2个月于外院手术治疗。未明确分类的先天性肺囊性疾病29例(30%),通过肺CT检查不能确诊,考虑"先天性肺囊性疾病",部分病例与坏死性肺炎很难鉴别,有2例经6、10个月随访肺部病变消失诊断为坏死性肺炎。39例手术治疗患儿中37例恢复良好,无反复下呼吸道感染。
结论
先天性肺囊性疾病的确诊需依靠影像学及病理检查,多数因呼吸道感染就诊时发现,以咳嗽发热为主要临床表现。诊断后尽早手术治疗,手术预后较好。
先天性肺囊性疾病是临床上比较少见的肺部先天性肺发育异常,包括先天性囊性腺瘤样畸形(congenital cystic adenomatoid malformation,CCAM)、肺隔离症、先天性肺囊肿及先天性大叶性肺气肿(congenital lobar emphysema,CLE)4类。这4类疾病临床变现相似,影像学上有时难以区分,需要病理确诊。由于肺部感染后可出现坏死及囊性变,短期内很难区分坏死性肺炎及肺囊性病继发感染。本研究对2010年1月至2019年3月于天津市儿童医院住院治疗的96例先天性肺囊性疾病病例进行回顾性总结。
对象和方法
一、对象
选择2010年1月至2019年3月于天津市儿童医院住院治疗的先天性肺囊性疾病患儿96例进行回顾分析,通过ICD-10编码进行病例检索,包括先天性肺囊性病、CCAM、肺隔离症、先天性肺囊肿、CLE 5种疾病编码。本研究获天津市儿童医院伦理委员会批准(L201908),符合患者豁免知情同意条件。
二、方法
1.临床数据收集:
通过ICD-10编码按疾病名称搜索病例,通过电子病历系统收集患儿的一般信息、临床表现、影像学检查、治疗和转归等信息。病例中的影像学报告需符合肺囊性病变表现,手术患儿需有病理报告确诊。
2.诊断依据[1,2,3,4]:
先天性肺囊性疾病的临床表现无明显特异性,可出现反复的肺部感染,有咳嗽、咳痰、发热、呼吸急促、发绀等症状,也可全无症状。在影像学的表现也有很多相似之处,可有单个或多个囊性影像,依靠肺CT明确诊断非常困难,需病理确诊。CCAM为肺部囊肿与腺瘤样畸形的混合,胸X线片可见边缘清楚的软组织密度影,内有囊性影像,CT可见单个或多个囊性病变,可伴有实性肿块,病理特点是以支气管腺瘤样增生替代正常的肺组织,肺呈块状、多囊性改变。肺隔离症是指部分肺组织与正常的支气管肺组织无交通,病变肺无正常肺功能,不接受肺动脉供血,异位的动脉通常来源于主动脉,肺隔离症分为叶内型和叶外型,胸X线片表现多为下叶内后方脊柱旁致密影,呈肿块样表现,增强CT可见隔离肺异常供血动脉,病变组织病理表现可有肺泡、支气管等呼吸上皮结构。先天性肺囊肿病理上分为支气管源性、肺泡源性、混合源性囊肿,支气管源性囊肿最多见,胸X线片及CT均可见气、液囊肿,病理上囊壁结构为支气管组织,内层有纤毛柱状上皮,外层可见散在软骨,囊内可见平滑肌束及纤维组织,可有单个或多个气囊肿、液气囊肿。CLE表现为肺叶过度充气扩张而基本不伴有肺泡间隔的破坏,外观肺体积增大,病变处充满空气,胸X线片及CT可见肺叶过度充气,内可见稀疏肺纹理,临近有压迫性肺不张,病理可见肺泡增大及局灶性肺气肿。
三、统计学处理
采用SPSS 18.0统计学软件进行数据处理,非正态分布计量资料采用M(范围)表示,计数资料以例(%)表示。
结果
一、一般情况
共96例先天性肺囊性疾病患儿,男50例(52%),女46例(48%),年龄2岁(4 d~13岁)。CCAM 40例(42%)、肺隔离症12例(13%)、先天性肺囊肿12例(13%)、CLE 3例(3%)、未明确分类的先天性肺囊性疾病29例(30%),通过肺CT检查不能明确诊断,考虑"先天性肺囊性疾病"。
7例患儿于胎儿期进行产前系统性超声检查发现肺发育异常,生后经肺CT诊断CCAM 4例、肺隔离症2例、先天性肺囊肿1例。4例患儿分别于生后3~10个月行手术治疗,术前均无呼吸道感染,术后病理诊断CCAM 2例、肺隔离症2例。另外3例因肺炎住院治疗,未手术。4例患先天畸形,分别为先天性肛门闭锁、房间隔缺损、主动脉缩窄及左肾缺如。
86例患儿因呼吸道感染就诊,有咳嗽、发热症状,10例患儿因查体发现肺部异常或呼吸困难就诊。既往反复肺炎6例,有3例为CCAM、3例为肺隔离症叶内型。合并喘息共15例,4例为术前有喘息,术后随访均无喘息发作。另有1例CCAM为术前无喘息,术后反复喘息发作,术后1年半复查CT示右肺中下叶支气管纤细狭窄,纤维支气管镜检查可见右下后基底段支气管闭塞。96例患儿中仅有4例行肺功能检查,1例肺隔离症患儿肺功能正常,3例潮气肺功能检查提示阻塞性通气功能障碍中-重度。共9例患儿行纤维支气管镜检查,6例提示支气管开口狭窄,3例支气管内可见大量分泌物阻塞。
二、CCAM
CCAM共40例,手术病理确诊30例,未手术10例。病变位于右肺下叶16例、左肺上叶3例、左肺下叶7例、右肺上叶9例、右肺中叶4例、左肺上下叶1例。择期手术28例,急症手术2例,死亡1例。择期手术中3例为产前系统超声检查发现肺内肿物,生后未发生呼吸道感染,分别于生后4、6、10个月手术治疗,其余25例为呼吸道感染就诊时发现肺内囊性肿物,抗感染治疗后手术治疗(1例右肺上叶CCAM手术患儿影像学检查见图1A、图1B,病理检查结果见图2A)。术前误诊3例,CT考虑先天性肺囊肿,术后病理确诊CCAM。急症手术2例,1例为1月龄男婴,因肺炎住院,CCAM病变位于左肺上叶及下叶,住院期间因进行性呼吸困难行急症手术,切除左肺上叶1~3段及左肺下叶6~10段,术后予机械通气治疗,感染未控制,8 d后死亡。另1例为CCAM合并膈疝行急症手术,术后恢复良好。30例手术病理免疫组织化学示上皮膜抗原、结蛋白、CD68、髓过氧化物酶等阳性。10例行核增殖抗原ki-67免疫组织化学,3例阳性,阳性率分别为1%、3%、10%。
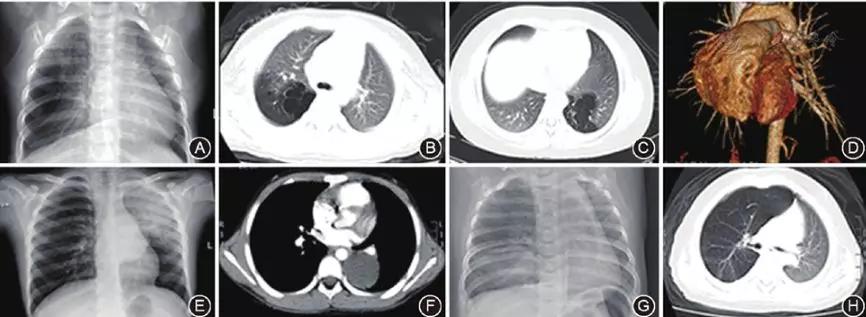
图1 先天性肺囊性疾病患儿手术前胸部影像学图片
A:右肺上叶先天性囊性腺瘤样畸形胸部X线片提示右肺中野外带类圆形囊性透亮影;
B:肺CT显示右肺上叶囊性透亮区;
C:左肺下叶肺隔离症胸部增强CT显示左肺下叶多发囊性透亮区;
D:增强CT血管重建影像;
E:左肺先天性肺囊肿胸部X线片提示左肺中上野片状实变影,左肺门类圆形软组织团块影;
F:增强肺CT显示左侧胸腔片状软组织密度影;
G:右肺上叶先天性大叶性肺气肿胸部X线片提示右肺上野透亮度增高,可见部分右肺组织跨纵隔向左侧胸腔疝入、右肺下野片状高密度影;
H:肺CT提示右肺气肿
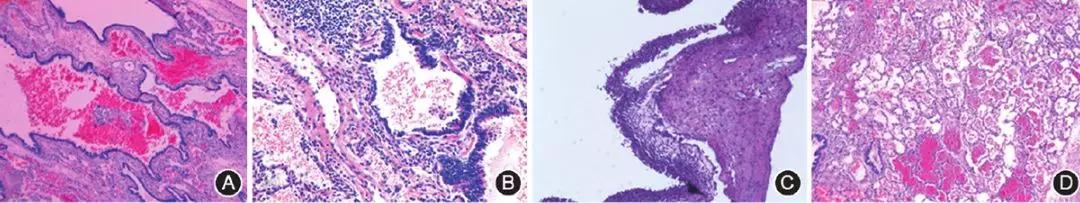
图2 先天性肺囊性疾病患儿镜下病理图片
A:先天性囊性腺瘤样畸形病变,囊壁见柱状上皮被覆(HE ×100);
B:肺隔离症病变,囊壁被覆柱状上皮,周围可见炎细胞浸润,结合临床符合肺隔离症(HE ×100);
C:先天性肺囊肿病变镜检,囊肿壁支气管上皮增生(HE ×100);
D:先天性大叶性肺气肿病变,部分肺泡腔过度充气,局部肺泡融合,肺泡内广泛出血(HE ×100)2例行纤维支气管镜检查,1例为CCAM术后反复喘息,气管镜下显示右下后基底段支气管闭塞。另1例为肺CT提示左肺上叶过度充气,左肺上叶支气管开口狭窄,气管镜检查显示左肺上叶固有段支气管狭窄,手术切除左肺上叶。30例手术患儿28例出院后恢复良好,无反复下呼吸道感染,有5例患儿因肺炎再次住院治疗,间隔最短1个月,最长3年。
CCAM未手术患儿10例,均因肺炎住院,9例患儿肺炎好转后出院。放弃治疗1例,为15日龄新生儿因呼吸困难就诊,入院后即予呼吸机辅助通气,3 d后家属放弃治疗。有2例出院后未再患肺炎,7例失访。
三、肺隔离症
肺隔离症共12例,均由肺增强CT确诊,病变位于左肺下叶8例、右肺中叶1例、右肺下叶3例。手术治疗4例,叶内型1例(病理结果见图2B),动脉来源于胸主动脉;叶外型3例,动脉来源均为腹主动脉。未手术8例,除1例12岁女孩为体检发现肺部异常外(肺CT见图1C、图1D),余均因肺炎住院后确诊。胎儿期发现肺发育畸形2例,分别于生后4、10个月手术治疗,术前未发生呼吸道感染。住院前患反复下呼吸道感染3例,2例手术治疗,均为肺隔离症叶内型;未手术的1例随访3年,共患2次肺炎。
4例肺隔离症术后患儿均恢复良好出院,2例随访无肺炎发生,1例术后2个月患肺炎,1例术后3年患肺炎。8例未手术患儿中5例失访,随访3例中无症状1例,再次患肺炎2例。
四、先天性肺囊肿
先天性肺囊肿12例,病变位于左肺上叶2例、右肺上叶4例、右肺中叶1例、右肺下叶5例。患儿均因肺炎住院,手术治疗4例(1例左肺上叶病变患儿影像学见图1E、图1F,病理结果见图2C)。胎儿期发现肺发育畸形1例,生后7年共患肺炎4次。放弃治疗1例,为1月龄患儿,CT考虑先天性肺囊肿合并纵隔肺疝,家属因经济原因放弃治疗。咯血1例,每日10余次,均为新鲜血丝或血块,抗感染治疗后好转。12例中9例失访,随访3例,1例于出院后10个月再次因肺炎住院治疗,2例随访半年无症状。
五、CLE
CLE 3例,病变位于左肺上叶1例、右肺上叶2例。手术1例(影像学检查见图1G、图1H,病理结果见图2D),该患儿生后4个月开始反复咳喘,多次CT检查提示右肺上叶气肿逐渐加重,左肺体积逐渐减小,1岁时手术切除右肺上叶。1例患儿因气胸住院,行胸腔闭式引流治疗,出院后2个月于外院手术切除右肺上叶。放弃治疗1例,为19日龄患儿因呼吸衰竭行呼吸机辅助呼吸,5 d后家属放弃治疗。
六、未明确分类的先天性肺囊性疾病
未明确分类的先天性肺囊性疾病共29例,所有患儿均因肺炎住院,CT提示有多发囊性病变但不能明确诊断。病变位于左肺上叶3例、左肺下叶6例、右肺上叶6例、右肺中叶2例、右肺下叶12例。19例患儿肺CT显示多发囊性透亮区及炎性实变,伴或不伴有胸腔积液,考虑先天性肺囊性疾病继发感染,无手术病理确诊具体类型。4例行纤维支气管镜检查,2例见支气管开口狭窄、气道畸形,1例见支气管开口明显增粗,1例见大量黄色分泌物阻塞支气管。气胸2例,行胸腔闭式引流治疗。29例患儿中3例分别于出院后2~11个月复查肺CT,炎症吸收但仍有囊性病变,未手术治疗。失访16例。
有10例患儿临床表现与肺CT不能区分是坏死性肺炎还是肺囊性疾病继发感染,需出院后继续随诊。患儿咳嗽发热时间长,病情进展迅速,伴有感染中毒症状,住院时间最长28 d,肺CT均显示有大片炎性实变及肺内空洞,治疗期间复查炎症好转,但空洞较前增大或变化不大,首先考虑坏死性肺炎,不能除外先天性肺囊性疾病继发感染。3例行纤维支气管镜检查,均可见大量黄色分泌物堵塞管腔,1例有支气管开口狭窄。2例患儿分别于出院后6、10个月复查CT,肺内空洞消失,诊断坏死性肺炎。另有3例患儿出院后短期复查肺内仍可见空洞,后失访。
讨论 先天性肺囊性疾病的发病率很低,多数没有具体的统计数据,CCAM的发病率为1/11 000~1/35 000[1]。CCAM、肺隔离症、先天性肺囊肿和CLE 4种疾病在临床表现上非常相似,可以完全无症状,或出现咳嗽、发热、咳痰等非特异性症状,在影像学上的表现也都有单个或多个囊性病变,需依据病理结果确诊。有极少数患儿生后就出现进行性的呼吸困难、面色发绀、咳喘,需急症手术,手术的风险非常高,本次研究中就有1例急症手术死亡病例。产前系统性超声(四维B超)检查在胎儿期就可发现肺部疾病,可在生后第一时间评估患儿肺内病变情况,是早期诊断非常好的方法。四维B超在胎龄17周左右即可发现肺内肿物[5],研究发现胎儿肺囊肿空腔大于3 cm及伴有胸腔积液、纵隔移位、羊水过多是生后出现症状的危险因素[6]。本次总结的病例中有7例为胎儿期发现肺部异常,因此产前超声检查还是非常必要的。CCAM某些类型和恶性肿瘤有关联[7],比如胸膜母细胞瘤、横纹肌肉瘤等,早期手术可减少恶变风险。本组患儿病理均未见肿瘤细胞。ki-67是反映细胞增殖活性的敏感指标,高表达可能和某些恶性肿瘤的预后相关,是淋巴结转移的预测因子[8]。本组有3例免疫组织化学ki-67阳性,阳性率最高10%,但高表达是否和恶变有关还需进一步研究。大多数患儿都是在出现呼吸道症状就诊时发现肺部囊性病变,此时已有感染,延误了最佳的治疗时机。因此早期诊断需重视肺CT检查。各类型肺囊性疾病在影像学上的表现有很多相似之处,只依靠CT没有病理非常难确诊,本组病例中就有3例CT考虑肺囊肿,病理结果为CCAM。肺囊性病变非常容易继发感染,比如叶内型肺隔离症、先天性肺囊肿等,影像上多数表现为大片炎性实变,在炎症吸收后才能显示囊性变,因此要定期复查,重视影像学变化。肺部感染后也可出现空洞性病变,如坏死性肺炎、肺脓肿等。由于病变从液化到坏死形成空洞的时间可以很短,甚至48 h CT就有明显变化[9],因此在没有既往影像学检查的情况下,确实很难确定是囊性病继发感染还是坏死性肺炎。坏死性肺炎最常见的病原是肺炎链球菌、金黄色葡萄球菌、腺病毒、肺炎支原体[10],临床上常表现为长期的发热、咳脓痰、胸痛、呼吸急促等,抗感染治疗预后较好,多数不需要外科手术可完全恢复正常,不留后遗症[11]。本组有2例患儿,最终通过6~10个月的随访,病变完全吸收,诊断坏死性肺炎。因此如果患儿发热咳嗽病程短,一般情况良好,早期影像学检查可见到囊性病变,考虑先天性肺囊性疾病的可能性大。如果发热时间很长,感染中毒症状重,合并胸腔积液、脓胸、脓气胸,CT可见到大片炎性实变及空洞性病变,考虑坏死性肺炎的可能性大,但需长期随访,定期复查CT非常重要。肺CT提示囊性病变需要鉴别朗格罕组织细胞增生症(langerhans cell histiocytosis,LCH)的肺损伤。单纯肺受累的LCH发病率很低,多见于成人,在儿童常见为多系统受累,如骨、皮肤、垂体损伤。唐晓蕾等[12]报道的14例儿童肺受累LCH均为多系统损伤。LCH早期肺CT可见实性小结节或网点状致密影,中期可见囊泡样改变,囊泡破裂可出现气胸,晚期可进展为肺纤维化。本组未明确诊断的患儿均无皮疹、骨损伤等其系统受累症状,肺CT无结节影,因此不考虑LCH。婴儿期发病肺囊性疾病的还需鉴别婴儿特有的间质性肺疾病,如基本突变引起的表面活性物质功能遗传性缺陷,但由于发病率极低,临床很少见到。先天性肺囊性疾病的治疗原则是明确诊断后早期手术切除病变。由于反复呼吸道感染会增加手术难度,加之择期手术并发症较急症手术明显减少[13],建议在婴儿期明确诊断后尽早手术[14]。有研究报道CCAM患儿即使没有症状,最早在3月龄病理就可见到炎症细胞浸润[15]。肺隔离症叶内型容易出现反复感染,本组既往反复肺炎的肺隔离症均为叶内型,因此建议发现后早期手术切除隔离肺组织[16]。由于肺隔离症叶外型感染发生率较少,是否必须手术治疗目前仍有争议,但是由于成人可见到很多肺隔离症患者,出现反复感染、咳血、胸痛等,因此也建议早治疗。胸腔镜下肺叶切除技术已经非常成熟,损伤较开胸手术小,恢复快,是目前推荐的手术方式。经动脉导管栓塞供血血管的介入治疗也取得了良好的效果[17],避免了开胸手术,但是由于缺乏长期的随访,可行性尚无定论。绝大多数患儿手术后恢复良好,生活不受影响。有研究对肺囊性疾病术后患儿进行长期随访,发现身体发育均无异常,大多数肺总量正常,耐力不受影响[18]。由于肺在生后2岁都有生长的能力[19],包括肺泡数量的增加和剩余肺肺泡体积的增大,因此肺叶切除后患儿肺功能也可恢复良好。但也有一些研究认为保守治疗也是可行的,Cook等[20]在一项119例先天性肺囊性病的研究中发现,57%的患儿选择保守治疗,预后很好,甚至有4例肺部病变消失,因此是否必须手术治疗目前还存在争议。本组有多例未手术患儿,目前生长发育良好,无反复肺炎,但随访时间最长3年,还需继续观察。关于先天性肺囊性疾病发病的分子生物学机制目前有很多研究,如成纤维细胞生长因子10信号放大可引起肺部细胞的过度增殖形成囊性变[7]、同源盒基因b5表达异常影响肺发育成熟等[21],尚无公认的机制。是否有明确的异常基因与之发病相关目前研究还很少,这也是以后进一步研究的方向。
#健康科普排位赛# #医师报超能团# #超能健康团#