
王蕾 马建民
视神经胶质瘤是一种原发于视神经胶质的肿瘤,根据其侵犯部位不同可以分为眶内视神经型、视交叉型、颅内弥散型,单纯眶内视神经胶质瘤临床少见,占眶内肿瘤的1% ~ 6%。本文主要就1例发生于3岁患儿的视神经胶质瘤的诊治情况进行报道,以期为读者了解和认识此病提供资料。
病例报道
患儿男性,3岁,主因左眼突出3个月,伴左眼眼球向内偏斜1个月就诊。3个月前患儿家长发现患儿左眼轻度突出,突出不明显故未予诊治;近1月来,患儿左眼突出加重,并出现眼球向内偏斜,故就诊于北京同仁医院眼肿瘤专科门诊。患儿自发病以来,无眼红、眼痛、流泪增多等症状。全身查体:神志清、精神可,心、肺、腹未见异常,皮肤未见异常。眼科查体:视力右眼0.5左眼0.1,眼压 右眼16mmHg,左眼22mmHg。左眼眼球突出,内斜约10度,运动受限,角膜透明,瞳孔直径约5mm,直接对光反射消失、间接对光反射存在,KP(-),眶压可;右眼未见异常。眼底检查:左眼视盘水肿,黄斑中心凹反光不清;右眼底未见异常。眼眶MRI扫描显示,左眼眶肌锥内一梭形肿块,来源视神经,边界清,大小约30X15mm,视神经管扩大,眼球受压变形;T1WI相示病变为稍高信号,不均匀,内可见低信号区,T2WI相示病变为高信号,肿物边缘可见低信号,增强扫描后可见肿物不均匀强化,中心低信号区未强化;右眼未见异常。考虑左视神经胶质瘤可能性大(图1)。患儿在全麻下行左眶内肿物切除术,术中见肿物来源视神经,视神经明显增粗,病理组织学证实为左视神经胶质瘤。术后请儿科和放疗科进一步诊治。随访至今4月余,未见复发及转移迹象。
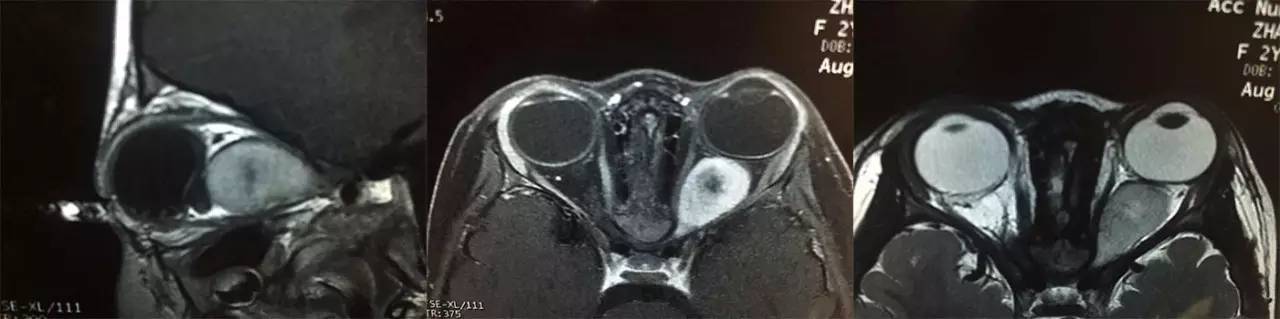
图1. 患儿术前MRI影像
讨论
视神经胶质瘤是来源于神经胶质的肿瘤,可发生于视神经的任何一段,并沿着视神经纵轴生长[1],是最常见的原发于视神经的肿瘤。根据肿瘤侵犯部位可分为眶内视神经型、视交叉型、颅内弥散型,单纯眶内视神经胶质瘤临床少见,占所有视神经胶质瘤的10%~30%[2],占眶内肿瘤的1% ~ 6%[3]。大约1/3的视神经胶质瘤仅局限于一侧视神经,而2/3的患者合并有视交叉、下丘脑、第三脑室底及视束的侵犯。
视神经胶质瘤发病无明显性别差异;在5岁以内发病的儿童占65%,10岁以内发病者占75%,20岁以内发病者占90%[4-6];平均年龄为5~6岁[7.8],发生于成人者少见。据最新统计,在国外约30%~50%的视神经胶质瘤患儿合并有神经纤维瘤病[1.7.9],而15%~20%的神经纤维瘤病患儿表现有视神经胶质瘤[10.11],多为双侧发病。国内报道的视神经胶质瘤却很少合并神经纤维瘤,本例患儿就没有发现神经纤维瘤病的体征。成人视神经胶质瘤多为恶性,预后较差。
视神经胶质瘤一般生长缓慢,肿瘤沿着视神经浸润生长,很少突破表面包膜向眶内侵犯。其自然病程多样,可静止、可进展,伴有神经纤维瘤的视神经胶质瘤为良性自限性肿瘤,患者病情长期稳定,甚至可见自然消退,而不伴有神经纤维瘤的视神经胶质瘤生长较快,具有侵袭性,预后差[7.12.13.14]。眶内视神经型胶质瘤典型的临床表现为早期出现视力逐渐下降、视野缺损,随着病变体积增大,出现眼眶占位性病变的特点,即进行性眼球突出、眼球运动受限、斜视等,故视力下降先于眼球突出出现。在婴幼儿期的视神经胶质瘤多表现为眼球震颤和斜视[15-17]。儿童出现视力下降时因其主诉不明确,往往在出现明显的眼球突出或因视力丧失导致废用性斜视时被家长发现才就诊,故儿童患者就诊时视力损伤已很严重甚至失明[7.18.19]。查体时可见患侧瞳孔扩大,直接对光反射消失、间接对光反射存在。肿瘤压迫可使眼底血管循环障碍,眼底检查可见患眼视乳头水肿或视神经萎缩。视神经的改变与肿瘤原发位置有关,肿瘤位于视神经前部则多发生视乳头水肿,并可伴有视网膜静脉扩张充血;原发部位在视神经后段,则原发性视神经萎缩多见。此外,长期视乳头水肿最终亦表现为视神经萎缩[20]。成人发病的视神经胶质瘤恶性程度高,70%~84%的患者常表现为突然视力下降,往往不伴有眼球突出[21-23],可出现视网膜中央静脉阻塞,虹膜新生血管性青光眼,甚至眼缺血综合征等并发症[15.24]。视交叉胶质瘤以双侧视力下降、视野改变多见,预后较差。弥散型胶质瘤可出现颅内压升高及肿瘤占位相应表现,但通常不具有特异性,因累及下丘脑而出现下丘脑损伤性症状,如性早熟、肥胖、多饮、多尿等内分泌异常,严重者可致死。
影像学检查对于肿瘤的发现、评估具有重要意义。视神经胶质瘤的影像学特点可以总结如下:(1)视神经梭形增粗、迂曲,或呈椭圆形肿块,与眶内脂肪分界清楚,瘤体内可见透亮区,为黏液池或囊变,CT表现为多发低密度影,可有点状钙化;(2)CT增强扫描呈中等均匀强化,冠状位可见视神经增粗,薄层像或骨窗像常见视神经管扩大,如果视神经管未见扩大诊断较困难,须借助MRI;(3)MRI对软组织的分辨力强、无放射性,是首选的检查方法,肿瘤最外周呈长T1、长T2信号,与脑脊液信号相似;肿瘤在T1相上与脑实质信号相比呈低信号或等信号,在T2相上呈高信号;增强扫描见多数肿瘤呈轻至中度均匀强化,部分肿瘤内部坏死或囊性变而不强化,少数胶质瘤可不强化[25.26]。当视神经胶质瘤同时累及眶内段、管内段及颅内段则可表现为哑铃形。
眶内视神经胶质瘤应与以下疾病相鉴别:(1)急性视神经炎:发病与感染、多发性硬化等有关,分为视神经乳头炎以及球后视神经炎,主要表现为急性或亚急性视力下降,视野缺损,以中心暗点及旁中心暗点为主,也可伴其他类型视野缺损;视神经乳头炎眼底可见视乳头水肿、视盘静脉充盈,而球后视神经炎的眼底多正常,但是有眼球转动痛。相对性瞳孔传入障碍是视神经炎最常见的瞳孔异常;MRI显示视神经增粗,但并不具有特异性[27]。应用糖皮质激素治疗有效[28]。(2)前部缺血性视神经病变(anterior ischaemic optic neuropathy, AION):多见于中老年人,表现为突发单眼无痛性视力下降、视盘水肿、与生理盲点相连的象限性视野缺损[29],视乳头水肿常于6~12周内消失,残留视盘苍白[12]。(3)视神经鞘脑膜瘤:占眶内肿瘤的4%~8%,中老年人发病多见,临床表现与视神经胶质瘤很相似,以视力下降以及眼球突出为主要表现,眼底检查可见视盘水肿,影像学检查对于二者的鉴别有很重要的意义。视神经鞘脑膜瘤常穿透硬脑膜形成偏心性肿块,如未穿透者,则表现为视神经一致性增粗,与视神经胶质瘤的梭形增粗不同。(4)眼眶横纹肌肉瘤:儿童时期最常见的原发性眶内恶性肿瘤,主要发生于10岁以下儿童,男性多于女性。起病急,进展迅速,主要临床表现为发展迅速的眼球突出及移位,早期即可出现眼球运动受限;横纹肌肉瘤恶性程度高,可侵犯周围眶壁,亦可向颅内蔓延,晚期可远处转移[30.31]。
WHO将视神经胶质瘤分为I、II两个级别。I级为低级别,为毛细胞性星形细胞瘤(pilocytic astrocytomas, PA),肿瘤生长缓慢、预后好,大多数伴有神经纤维瘤的视神经胶质瘤为此型;粘液性纤维型星形细胞瘤(pilomyxoid astrocytomas, PMA)是PA的一种亚型,相对少见,但多见于婴幼儿,肿瘤多位于视交叉及下丘脑部位,PMA呈进展性生长,容易复发,预后不好[32.33]。两者的病理学特点亦有所不同。在形态学上,PA由双极细胞以及多极细胞组成,双极细胞紧密连接,形成罗森塔尔纤维(一种紧密排列的神经胶质纤维)和嗜酸性颗粒小体,多极细胞疏松排列,含有微囊结构;而PMA则表现为在粘液背景中可见肿瘤细胞分布,细胞以血管为中心分布,缺乏罗森塔尔纤维和嗜酸性颗粒小体。近年来有报道发现PMA与PM之间可相互转化。恶性程度高的视神经胶质瘤为高级别即II级PA,又称为多形性成胶质细胞瘤,多见于成人发病的视神经胶质瘤[12]。
关于视神经胶质瘤的治疗,应根据患者的年龄、发病部位、临床表现及影像学检查进行综合分析,制定个性化治疗方案。部分患者的肿瘤稳定,呈静止状态,尤其是伴有神经纤维瘤的患者,大多无临床症状、视力保留较好,且肿瘤较小、局限于眶内,多采取定期观察或单纯放疗[8.10.34]。然而多数视神经胶质瘤逐渐增长,需要治疗。在随访过程中若出现视力不断减退、眼球突出加重、影像学检查肿瘤进展,如肿物出现增大、囊性变、外生性等,便应尽早切除[35]。放射治疗可以控制肿瘤的生长,标准总剂量超过50Gy[36],但是由于副作用明显,传统的放射治疗方法现今很少应用[37];近年来,立体定位放射治疗、质子放射治疗、以及短距离放射治疗等新兴的治疗手段,由于肿瘤接受的剂量更精确、可控,副作用少等优点,得以广泛应用。此外,对于复发的神经胶质瘤放射治疗的效果好。对于局限于一侧视神经的肿瘤,为保护眼球、阻止肿瘤向视交叉蔓延,手术完全或部分切除肿瘤的重要性逐渐受到关注[35]。此外,手术后进行组织病理学检查,不仅可以明确诊断,还可以根据肿瘤的病理类型制定后续的治疗方案。对于疑有视神经管或颅内蔓延者则应采取手术、放疗、化疗或联合治疗[8]。近年来一致的观点认为,对于全身性或者进展性视神经胶质瘤,尤其是≤5岁的婴儿患者,预后较差,常采用化疗作为一线治疗[1.4.6.38.39],最常用的化疗方案是COPP方案(环磷酰胺+长春新碱+甲基苄肼+泼尼松龙)或顺铂+卡铂方案[7]。但是一些研究显示,化学治疗只能延迟了疾病的进展,而不能使患者的视力得到改善[40-42]。
对于儿童发病的视神经胶质瘤,五年生存率约为52%~75%[43-45]。大多数研究认为,视神经胶质瘤的预后与患儿年龄、性别、发病部位、以及是否伴有神经纤维瘤病等有关。年龄≥10岁、男性、眶内视神经胶质瘤、伴有神经纤维瘤病的患儿预后更好[7.45]。婴幼儿期(≤1岁)发病的视神经胶质瘤更具有侵袭性,对视力的损伤更严重,临床症状更明显,因此预后更差[7.45.46]。眶内视神经胶质瘤的死亡率为21%,而视交叉受累时死亡率上升至58%[47]。21%的患者会出现复发或肿瘤进展[48]。成年人发病的视神经胶质瘤恶性程度高,进展快,患者通常在发病后6~12个月内死亡[23]。
本例患儿发病年龄小,尽管家长发现患儿眼球突出3个月,并逐渐出现斜视,一般情况下视力下降先于眼球突出,故推测患儿实际发病时间更长,只是未引起家长注意而已。MRI上可见肿瘤体积大,来源视神经,视神经管增粗,肿瘤内部出现囊性变,这些均为视神经胶质瘤进展的临床表现。综合分析后行手术切除肿物,病理组织学结果证实为视神经胶质瘤。此病例提示临床医生,对于眼球突出的患儿,诊断时要考虑视神经胶质瘤的可能,对于瘤体体积较大并呈进展状态的视神经胶质瘤,建议切除肿瘤,根据手术后病理组织学结果辅以放疗/化疗,以期改善患儿预后。
参考文献
[1]Jahraus C D, Tarbell N J. Review: optic pathway gliomas. Pediatr Blood Cancer [J]. 2006, 46: 586-596.
[2]Paulis D D, Nicosia G, Taddei G, et al. Intracranial aneurysms and optic glioma – an unusual combination: a case report [J]. Journal of Medical Case Reports, 2016, 10(1):1-4.
[3]宋国祥.眼眶病学[M].北京:人民卫生出版社,2010:301~312.
[4]Massimi L, Tufo T, Di R C. Management of optic-hypothalamic gliomas in children: still a challenging problem [J]. Expert Review of Anticancer Therapy, 2007, 7(11):1591-1610.
[5]Chung E M, Specht C S, Schroeder J W. From the archives of the AFIP: Pediatric orbit tumors and tumor like lesions: neuroepithehal lesions of the ocular globe and optic nerve [J]. Radiographics, 2007, 27: 1159-1186.
[6]Opocher E, Kremer L C M, Da Dalt L, et al. Prognostic factors for progres- sion of childhood optic pathway glioma: a systematic review [J]. Eur J Cancer , 2006,42: 1807–1816.
[7]Varan A, Batu A, Cila A, et al. Optic glioma in children: a retrospective analysis of 101 cases [J]. American Journal of Clinical Oncology, 2012, 36(3): 287-92.
[8]Goodden J, Pizer B, Pettorini B, et al. The role of surgery in optic pathway/hypothalamic gliomas in children [J]. Journal of Neurosurgery Pediatrics, 2014, 13(1): 1-12.
[9]Arul G S. P.A. Piazzo, D.G. Poplack, Principles and Practice of Pediatric Oncology, 5th ed [J]. Surgical Oncology, 2006, 15(1): 57-57.
[10]Thiagalingam S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients [J]. Ophthalmology, 2004, 111(3):568-577.
[11]Mikaeloff Y, Chaix Y, Grill J, et al. Optic pathway gliomas in neurofibromatosis type 1. A longitudinal survey of 30 children in two multidisciplinary consultations [J]. Archives De Pediatrie, 2002, 9(8): 797-804.
[12]Shapey J, Danesh-Meyer H V, Kaye A H. Diagnosis and magement of optic nerve glioma [J].Journal of Clinical Neuroseience, 2011, 18: 1585-1591.
[13]Deliganis A V, Geyer J R, Berger M S. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen Disease) in childhood optic glioma [J]. Neurosurgery, 1996, 38(6): 1118-1119.
[14]Astrup J. Natural history and clinical management of optic pathway glioma [J]. British Journal of Neurosurgery, 2003, 17(17): 327-335.
[15]Miller NM. Primary tumours of the optic nerve and its sheath [J]. Eye, 2004, 181026–181037.
[16]Wilhelm H. Primary optic nerve tumours [J]. Curr Opin Neurol, 2009, 22:11–18.
[17]Listernick R, Charrow J, Greenwald M, et al. Natural history of optic pathway tumours in children with neurofibromatosis type 1: a longitudinal study [J]. Pediatr, 1994, 125: 63–66.
[18]Zeid JL, Charrow J, Sandu M, et al. Orbital optic nerve gliomas in children with neurofibromatosis type 1 [J]. J AAPOS 2006, 10: 534–539.
[19]King A, Listernick R, Charrow, et al. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome [J]. Am J Med Genet A, 2003, 122: 95–99.
[20]Avery R A, Fisher M J, Liu G T. Optic pathway gliomas [J]. Journal of neuro-ophthalmology: the official journal of the North American Neuro-Ophthalmology Society, 2011, 31(3): 269-278.
[21]Hartel PH, Rosen C, Larzo C, et al. Malignant optic nerve glioma (glioblastoma multiforme): a case report and literature review [J]. W V Med J, 2006, 102: 29–31.
[22]Wabbels B, Demmler A, Seitz J, et al. Unilateral adult malignant optic nerve glioma [J]. Graefes Arch Clin Exp Ophthalmol, 2004, 242: 741–748.
[23]Dinh T T, Wang Y Y, Rosenfeld J V, et al. Glioblastoma of the optic chiasm [J]. J Clin Neurosci , 2007, 14: 502–505.
[24]Liu GT. Optic gliomas of the anterior visual pathway [J]. Curr Opin Ophthalmol, 2006, 17: 427-431.
[25]Pepin S M, Lessell S. Anterior visual pathway gliomas: The last 30 years [J]. Seminars in Ophthalmology, 2009, 21(3): 117-124.
[26]Lee A G. Neuroophthalmological management of optic pathway gliomas [J]. Neurosurgical Focus, 2007, 23(5): E1.
[27]钱志敏, 云丽霞, 周爱春, 等. 视神经炎的磁共振成像与临床观察 [J]. 中国实用眼科杂志, 2001, 19(2): 155-157.
[28]蓝育青, 张驰, 夏朝霞,等. 急性视神经炎临床特征和疗效分析 [J]. 国际眼科杂志, 2008, 8(9): 1935-1937.
[29]Atkins E J, Bruce B B, Newman N J, et al. Treatment of Nonarteritic Anterior Ischemic Optic Neuropathy [J]. Survey of Ophthalmology, 2010, 55(1): 47-63.
[30]于潇菡, 侯世科. 眼眶横纹肌肉瘤诊断的研究进展 [J]. 医学研究杂志, 2008, 37(1):91-93.
[31]顼晓琳. 眼眶横纹肌肉瘤的临床研究进展 [J]. 国外医学:眼科学分册, 2002, 26(6): 372-376.
[32]Cottingham SL, Boesel CP, Yates AJ. Pilocytic astrocy- toma in infants: A distinctive histological pattern [J]. J Neuropathol Exp Neurol, 1996, 55: 654.
[33]Chikai K, Ohnishi A, Kato T, et al. Clinico-pathological features of pilomyxoid astrocytoma of the optic pathway [J]. Acta Neuropathologica, 2004, 108(108): 109-114.
[34]Grill J, Laithier V, Rodriguez D, et al. When do children with optic pathway tumours need treatment An oncological perspective in 106 patients treated in a single centre [J]. European Journal of Pediatrics, 2000, 159(9): 692-696.
[35]Walker D A, Liu J, Kieran M, et al. A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method [J]. NEURO-ONCOLOGY, 2013, 15(4): 462-468.
[36]Montgomery A B, Griffin T, Parker R G, et al. Optic nerve glioma: The role of radiation therapy [J]. Cancer, 1977, 40(5): 2079-2080.
[37]Cappelli C, Grill J, Raquin M, et al: Long-term follow up of 69 patients treated for optic pathway tumours before the chemotherapy era [J]. Arch Dis Child, 1998, 79:334–338.
[38]Silva MM, Goldman S, Keating G, et al. Optic pathway hypothalamic gliomas in children under three years of age: the role of chemotherapy [J]. Pediatr Neurosurg, 2000, 33: 151–158
[39]Nicolin G, Parkin P, Mabbott D, Hargrave D, Bartels U, Tabori U, et al: Natural history and outcome of optic pathway glio- mas in children [J]. Pediatr Blood Cancer, 2009,53:1231–1237.
[40]Jr D H M, Cohen M E, Friedman H S, et al. Carboplatin is effective therapy for young children with progressive optic pathway tumors: a Pediatric Oncology Group phase II study [J]. Neuro-oncology, 2000, 2(4): 213-220.
[41]Moreno L, Bautista F, Ashley S, et al. Does chemotherapy affect the visual outcome in children with optic pathway glioma A systematic review of the evidence [J]. European Journal of Cancer, 2010, 46(12): 2253-2259.
[42]Laithier V, Grill J, Deley M L, et al. Progression-Free Survival in Children With Optic Pathway Tumors: Dependence on Age and the Quality of the Response to Chemotherapy—Results of the First French Prospective Study for the French Society of Pediatric Oncology [J]. Journal of Clinical Oncology, 2003, 21(24): 4572-4578.
[43]Khafaga Y, Hassounah M, Kandil A, et al. Optic gliomas: A retrospective analysis of 50 cases [J]. International Journal of Radiation Oncology Biology Physics, 2003, 56(3): 807-812.
[44]Maryam Fouladi , Wallace D, et al. Survival and functional outcome of children with hypothalamic/chiasmatic tumors [J]. Cancer, 2003, 97(4): 1084-1092.
[45]Yong A, Cho B K, Kim S K, et al. Optic pathway glioma: outcome and prognostic factors in a surgical series [J]. Child S Nervous System, 2006, 22(9): 1136-1142.
[46]Sylvester C L, Drohan L A, Sergott R C. Optic nerve gliomas, chiasmal gliomas and neurofibromatosis type 1 [J]. Curr Opin Ophthalmol, 2006; 17: 7–11.
[47]Alvord Jr E C, Lofton S. Gliomas of the optic nerve or chiasm. Outcome by patients’ age, tumour site and treatment [J]. JNeurosurg, 1988;68: 85–98.
[48]Dutton J J. Gliomas of the anterior visual pathway [J]. Surv Ophthalmol, 1994;38: 427–452.