遗传性肾脏病是儿童青少年终末期肾衰竭的首要病因、成人终末期肾衰竭的第五大病因。据报道,大约30%的病例有肾脏疾病家族史,揭示了遗传因素在肾脏疾病中的重要作用。遗传性肾脏疾病是由基因突变所致,并按孟德尔定律遗传子代的一组肾脏疾病。建立基因诊断对肾脏病诊疗有重要意义,因为可以合理进行治疗选择、科学预测预后,使患者最大程度免于接受侵入性诊断程序,如肾活检,并指导遗传咨询。目前有许多基因检测方法(如靶向测序、微阵列、基因检测包、全基因组方法等),可根据各种因素选择最合适的诊断测序方法。
这里举出一个实例。患儿,男,11岁,全外显子组测序和生物信息学分析发现在COL4A5基因中有一个致病性的大截短变异,诊断为Alport综合征。除了建立Alport综合征的分子诊断外,变异体分析还有助于对患者临床过程的深入认识,其*功中**能缺失变异体与早期肾功能衰竭、听力损失和眼部异常相关。COL4A5基因的大截短变异与移植后抗肾小球基底膜疾病的风险增加有关。此外,基因诊断可能会告知高危家庭成员的同种异体移植物供体选择,因为患有X连锁Alport综合征的男性母亲通常不建议作为供者。此外,建议应用抑制RASS的药物(如ACE*制剂抑**或ARB)保守治疗蛋白尿。最后,基因诊断还可以帮助确定特定疾病的支持群体,为受影响的个人及其家庭提供心理社会支持,指导患者进行临床药物试验、注册和利用其他相关资源。
【1】遗传性肾脏病、基因检测方式和诊断获益
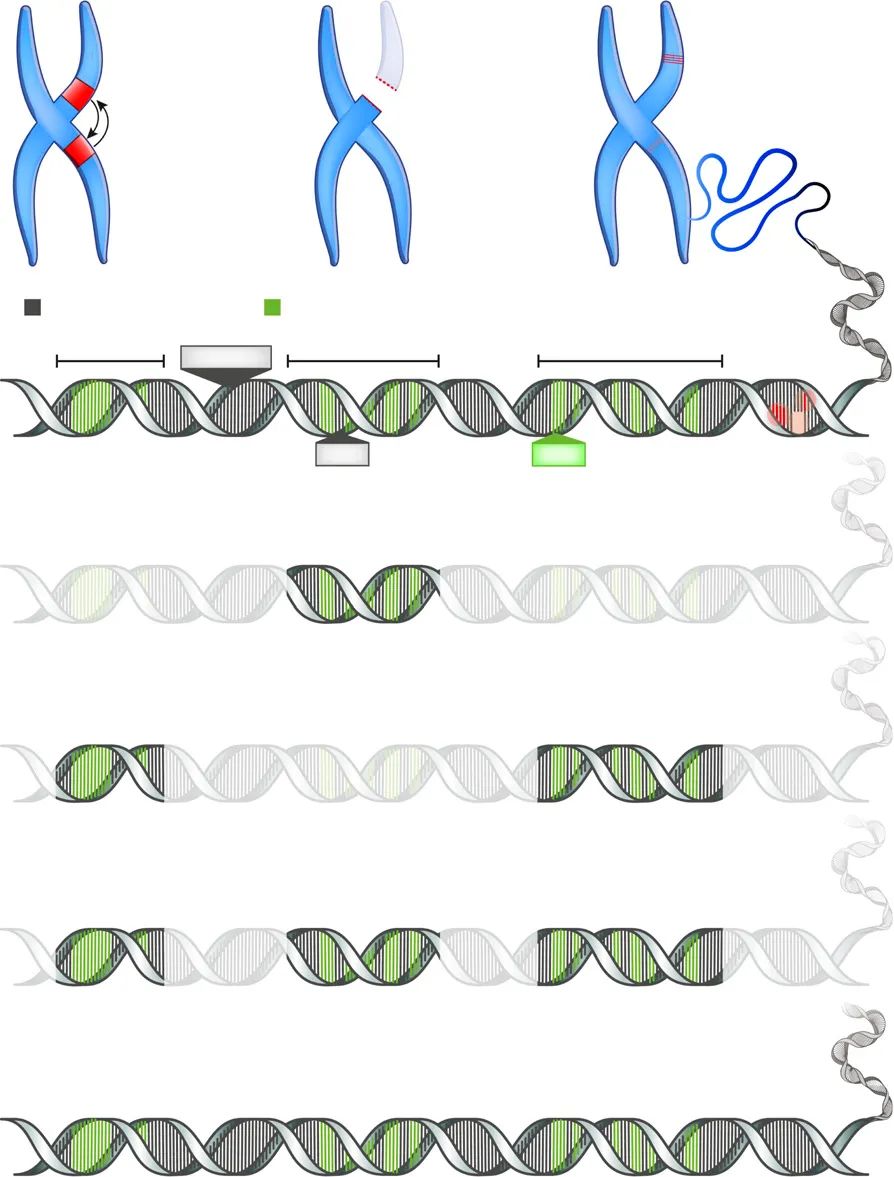
人类基因组在个体之间差异很大,遗传序列的变化在图1中进行了总结,包括单核苷酸变异、小片段插入缺失(INDEL)、拷贝数变异、染色体不平衡和重排。
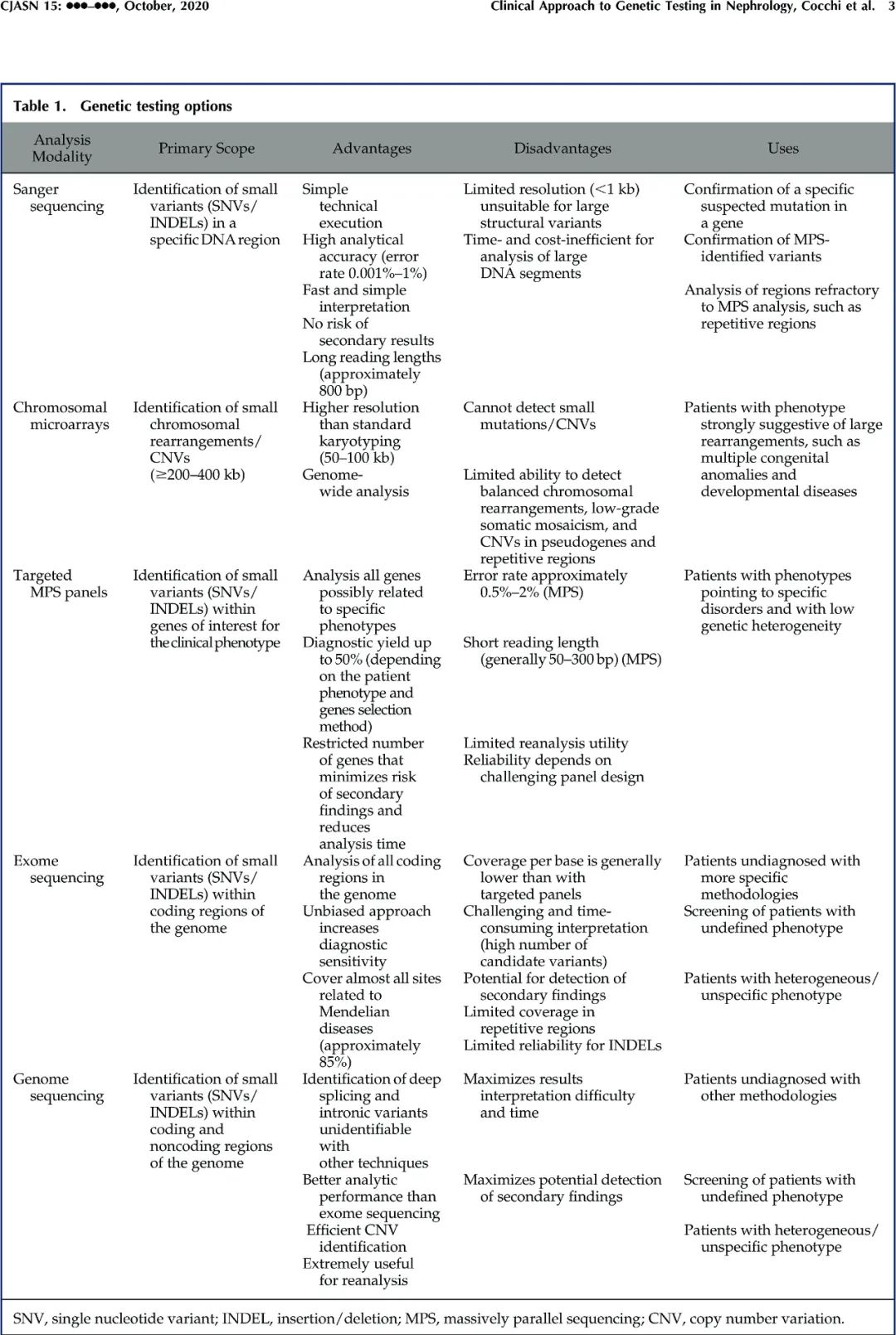
这些变异可能导致孟德尔病。最近的一项调查确定了625种与肾脏疾病相关的孟德尔疾病,而随着大规模平行测序的广泛应用,基因-疾病关联的数量继续增长,这一技术进步提高了基因组测序的通量。重要的是,诊断率随变异类型和检测方式的选择不同而不同。在图1和表1总结了不同测序方法的技术和临床方面以及它们各自的优缺点。
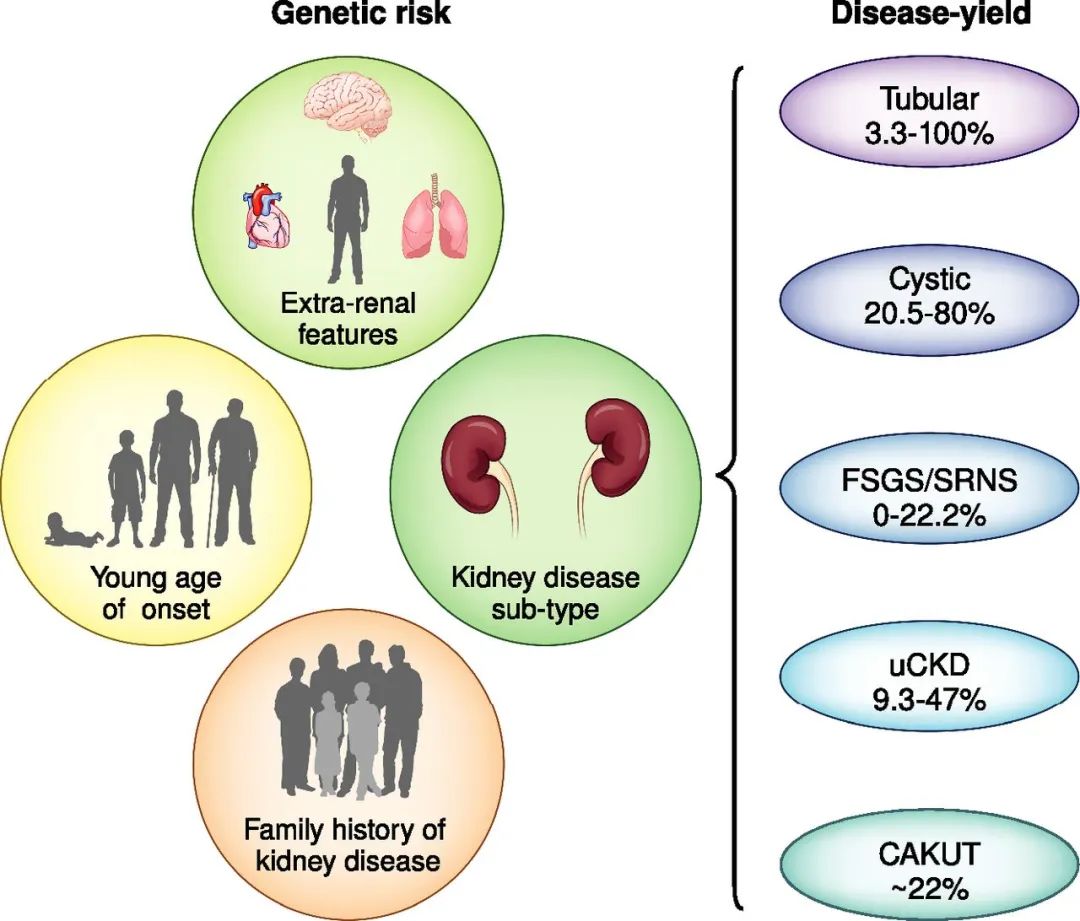
【2】遗传风险的临床决定因素及其对基因检测类型的影响
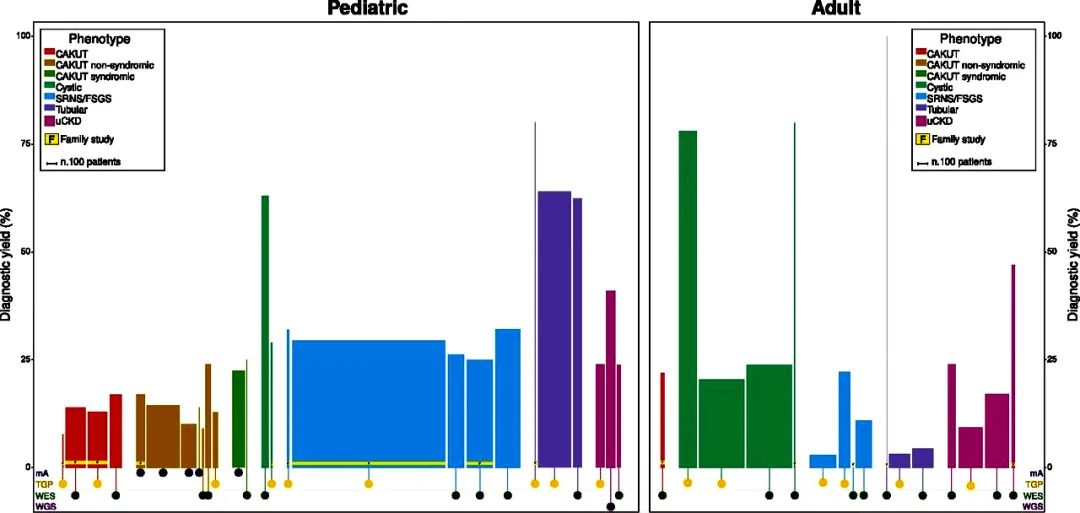
图中总结了肾脏病患者遗传特征。发病年龄小、肾脏疾病家族史、肾外病变的存在都是遗传性肾脏病的预后预测因素。各种遗传性肾脏病基因检测的诊断率也不尽相同。
【3】从基础至临床:COL4A5基因插入突变致Alport综合征一家系报告
先证者,男,11 岁8 个月,拟诊肾病综合征入院。
5 岁时无明显诱因下出现眼睑、面部浮肿,后渐及双下肢,伴尿量减少,当时无尿频、尿急、尿痛,无腰背部疼痛,无头痛呕吐,无抽搐,无发热咳嗽等。在当地医院诊断为肾病综合征,予以泼尼松治疗(具体不详),尿蛋白++后出院。之后患儿病情反复,在多家医院住院治疗,曾予以吗替麦考酚酯、环磷酰胺、他克莫司等治疗,尿蛋白反复++ ~+++。
于半年前停用激素及免疫*制剂抑**。
两周前患儿感冒后浮肿再发,呈咳嗽、咳痰,伴气喘,无发热、抽搐,无呕吐、腹泻,无头痛,无尿红,无少尿,拟诊肾病综合征入院。
入院体格检查:体温36.5℃,心率114次/min,呼吸24次/min,血压134/ 75 mmHg;神清,精神可,柯兴貌,颜面部、双下肢无浮肿,咽无充血,双侧扁桃体无明显肿大;双肺呼吸音粗,未闻及干湿啰音;心律齐,未闻及明显病理性杂音;腹稍膨,肝、脾肋下未及,移动性浊音阴性,双肾区叩痛阴性;神经系统检查无异常。双耳中度神经性听力障碍,双眼视力无异常。
实验室检查:尿常规潜血++,尿蛋白++++,红细胞32 个/μL。血超敏C反应蛋白<1 mg/L,血常规白细胞计数12.42×10^9 /L,中性粒细胞绝对值7.86×10^9 /L,血红蛋白164 g/L,红细胞压积50.2 %,血小板计数264×109 /L;红细胞沉降率25 mm/h;血白蛋白28. 3 g/L,白球比例1.29,前白蛋白0.35 g/L,三酰甘油5.64 mmol/L,胆固醇9.41 mmol/L,淀粉酶35.0 U/L;抗核抗体均无异常;TORCH抗体均无异常;T细胞亚群CD3 25 .12 %,CD4 4 .68 %,CD8 15 .02 %,CD4/CD8 0 .31;乙肝定性、HIV梅毒、丙肝均为阴性;结核抗体(38 KDa)阴性,结核抗体(16 KDa)阴性,结核抗体(LAM)阴性。24 小时尿量1300 mL,微量总蛋白3062.7 mg/L,24 小时尿蛋白定量3 981.5 mg;尿蛋白/ 尿肌酐:1.83。尿培养:无菌生长。
胸片未见明显异常征象。
腹部B超示胆囊结石。双肾B超示双肾皮质回声增强。
骨龄评估示骨龄落后。心电图示正常。
双肾磁共振成像(MRI)示双肾外形丰满,右肾下方脂肪间隙异常信号,渗出性改变可能,提示胆囊结石可能,两侧少量胸腔积液。
肾穿刺:组织HE染色示肾小管发育不良,泡沫细胞增多,出现幼稚肾小球;荧光染色示肾小球基底膜IV型胶原缺乏。
电镜检查示基底膜变薄,厚度约200 nm,系膜增生,系膜区有少量电子致密物沉积。患儿肾组织病理表现见下图。
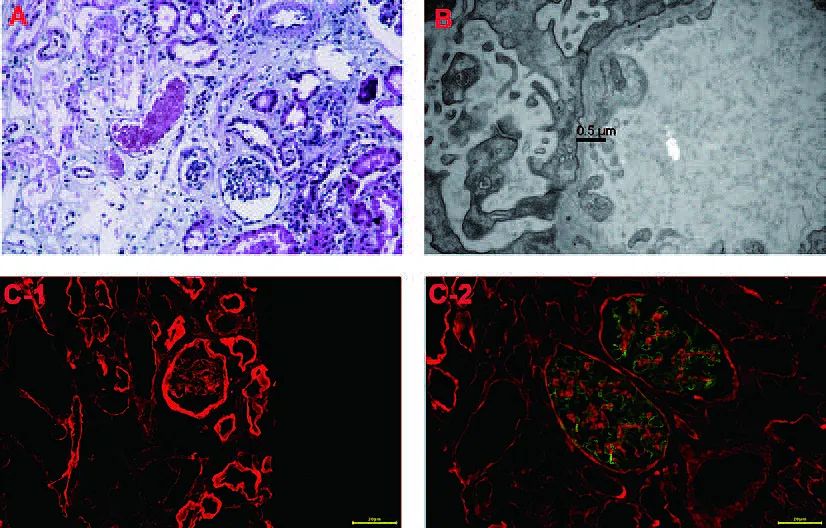
A:HE染色× 100 ;B:透射电子显微镜照片;C- 1 :先证者肾小球中胶原蛋白的荧光染色;C- 2 :正常对照中胶原的荧光染色(C- 1 和C- 2 红色荧光代表Ⅱ型胶原蛋白和绿色荧光代表Ⅳ型胶原蛋白)
为鉴定该家系的致病突变,经医学伦理审核及家属知情同意进行全外显子组测序和生物信息学分析。结果在先证者的COL4A5 基因发现TTCT 插入突变(c. 41 _ 42 dup TCTT),并进一步经Sanger 测序证实。依照ACMG临床实践指南该突变鉴定为致病。使用Integrative Genomics Viewer 软件分析显示,该插入突变引起移码突变,移码突变导致自插入之后的第13 个氨基酸残基开始发生改变,且在第40 个氨基酸残基处出现终止密码子,导致蛋白表达提前终止。见图2。
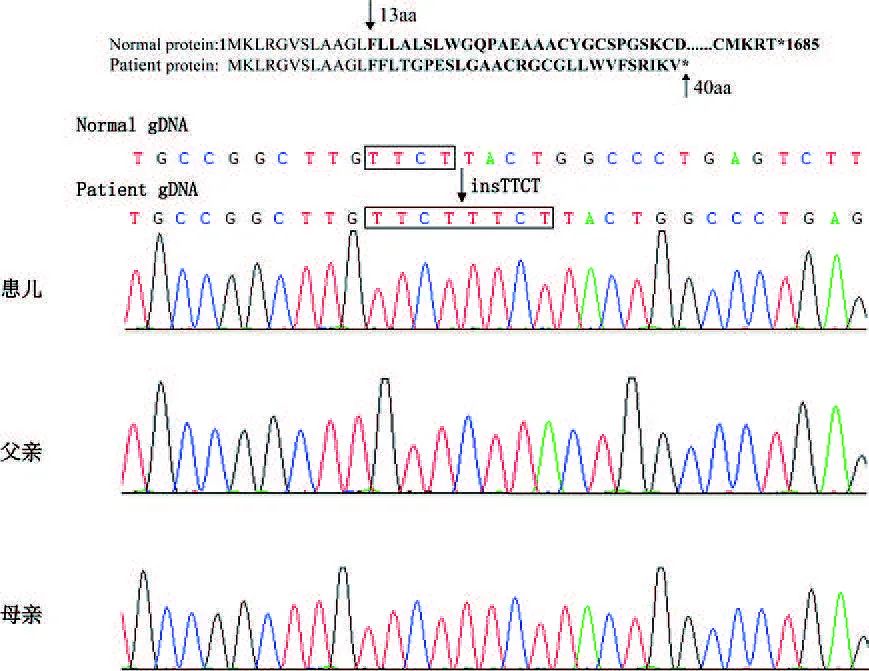
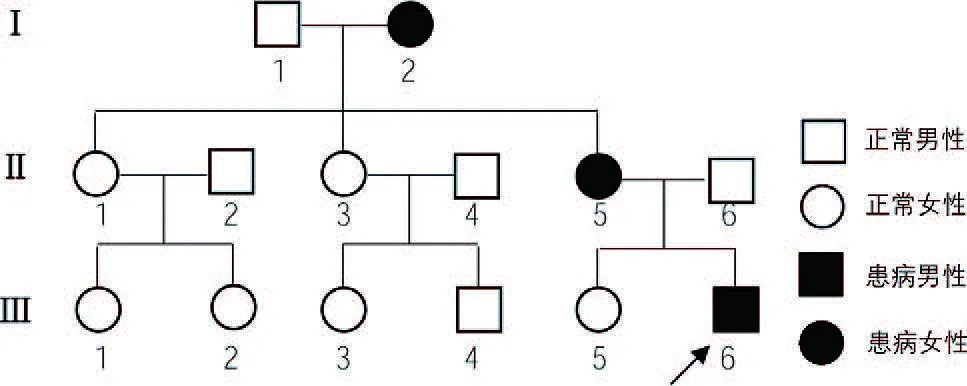
先证者的其他家系成员均采用Sanger 测序进行同一可疑致病变异的检测。患儿母亲(II 5)和外祖母(I 2)均携带该突变基因,分别在35 岁和34 岁时达到终末期肾病(end stage renal disease,ESRD),且两者都有听力受损,但未发现眼部异常。该家系的其他成员中未发现同样的致病变异,表明该变异与家系患病成员存在共分离关联(图3)
目前,尚无根治的方法。临床主要通过减少尿蛋白、延缓肾纤维化等方式来延缓肾衰竭的发生。根据基因检测结果,建议应用抑制RASS的药物(如ACE*制剂抑**或ARB)保守治疗蛋白尿。
【4】遗传性肾脏病研究领域的国内重要进展
遗传性肾脏病是儿童青少年终末期肾衰竭的首要病因、成人终末期肾衰竭的第五大病因。遗传性肾脏病是儿童和青少年慢性肾病的主要病因之一,20%的25岁以下慢性肾病患者其病因为单基因突变所致遗传性疾病,而在终末期肾衰竭儿童中与遗传相关的原发肾脏疾病约占70%甚至更高。复旦大学附属儿科医院徐虹教授带领的多中心团队,绘制出了首个中国儿童肾脏病基因突变图谱,相关研究成果以题为“基于多中心注册登记系统的1001例儿童肾病疾病致病基因谱系研究”的文章发表在Clinical Genetics杂志,引起国际领域高度关注。该研究依托“中国儿童遗传性肾脏疾病数据库”首批来自全国17个省23家医疗机构儿童肾脏专科共1001例不同类型儿童肾脏病的临床表型及基因测序数据。研究团队通过表型与基因型交互验证的数据分析方法,使42.1%的患儿明确了致病基因的突变,显著提高了儿童遗传性肾脏病的分子诊断阳性率。该致病基因图谱凸显了中国人群在不同类型肾脏疾病致病基因的谱系特征,其公布有望改变临床工作者对我国儿童遗传性肾脏病诊断难度大、救治手段匮乏的观念,并为我国罕见及遗传性肾脏疾病的诊断和治疗赋能。
既往笔记
搭建精准医学平台助力儿童遗传性肾病诊疗