2016年,全球医药市场规模超过1万亿美元,医药研发成本不断攀升,小分子新药获批数量日益降低,抗肿瘤药物、孤儿药研发势头依旧不减。随着生物药专利悬崖逐渐显现,生物类似药崭露头角。遴选了2016 年上市的部分重磅新药,盘点了抗感染、抗肿瘤、中枢神经系统等领域的新药研发进展,回顾了2016 年国内药学领域的相关热点事件。
随着新兴市场国家经济飞速发展、中产阶级迅速扩张,全球医药产业规模不断扩大,据艾美仕市场研究公司(IMS Health)数据显示,2015年全球医药市场规模已超过1万亿美元,未来5年的年增长率在4%~7%,预计在2020年,全球医药市场规模将达到1.4万亿美元,全球医药产业陷入低速增长期。据推测,未来5年全球专利药面临1780 亿美元的损失。与此同时,医药研发成本不断攀高,小分子新药获批数日益降低,生物新药和生物类似药的研发成果逐渐显现。
根据美国美国食品药品监督管理局(FDA)官网显示,截至2016年12月27 日,FDA 下属药品评价和研究中心(CDER)共批准了22 个新分子实体(new molecular entities,NMEs),包括15 个化学小分子药物和7 个生物新药。2016年获批的新药数较2015年明显下降(2015年获批45个),但生物新药获批比例有所提高,由27%提高到32%。此外,2016年,FDA还批准了11个新组合制剂(new drug combinations)。本文遴选2016年上市的部分重磅新药,盘点抗感染、抗肿瘤、中枢神经系统领域新药的研发进展,并回顾2016年国内药学领域的相关热点事件。
抗感染药物再崛起
抗感染药物在20世纪初期显露头角,在20世纪70年代占据了药物市场1/3的份额。近几十年来,随着人们对抗感染药物机理的深入研究,使得绝大部分的感染性疾病有了稳定且疗效显著的治疗方案,新型抗感染药物凤毛麟角。然而2016年,抗病毒的新药或新组合制剂频频获批,如抗炭疽热药物Anthim,抗艰难梭菌感染药物Zinplava,抗丙肝药物Zepatier、Epclusa、Odetsey、Descovy,抗乙肝药物Vemlidy等。而这一领域最大的赢家,莫过于吉利德科学公司(Gilead Sciences, Inc.)。
丙肝全基因型治疗药物:Epclusa
2016年6月和7月,吉利德开发的泛基因型丙肝鸡尾酒疗法药物Epclusa在美国和欧盟获批用于治疗全部6种基因型丙肝。
全球约有1.8亿人感染丙肝病毒。在美国,基因型1丙肝是最常见的丙肝类型;然而,在全球范围内,大约75%的美国人为基因1 型丙肝,20%~25%为基因2或3型。目前,尽管丙肝的临床治疗已经成熟,但仍有许多患者亟需一种简易高效的泛基因型丙肝药物,尤其是针对极为难治的3型HCV患者。
Epclusa是全球首个也是唯一一个全口服、泛基因型、单一片剂的丙肝治疗方案,也是获批治疗基因型2和基因型3丙肝的首个单一片剂方案(不需要联合利巴韦林)。Epclusa是索非布韦(Sofosbuvir)和新药Velpatasvir 的固定复方口服片剂(图1、图2)。
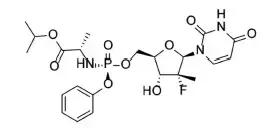
图1 Sofosbuvir 结构式
图2 Velpatasvir 结构式
Sofosbuvir是一种核苷类似物聚合酶NS5B *制剂抑**,Velpatasvir则是一种泛基因型NS5A*制剂抑**。
Epclusa 的获批,是基于4 项III 期ASTRAL临床研究的积极数据,这些研究在全部6种基因型(GT-1,-2,-3,-4,-5,-6)丙肝群体中评估了Epclusa 的疗效和安全性。数据显示,该鸡尾酒疗法针对所有6种基因型丙肝全部有效,包括伴有代偿性和失代偿性肝硬化患者群体。4项研究中,1035例泛基因型丙肝患者用药12 周后,治愈率达到了98%。
目前,吉利德的另两款丙肝药物已帮助吉利德占据了超过85%的市场份额。而Epclusa在提供高临床治愈率的同时有望简化丙肝治疗过程,消除检测患者丙肝基因型的必要性。业界对Epclusa的商业前景非常看好,认为这款泛基因型丙肝鸡尾酒药物将进一步巩固吉利德在丙肝领域的绝对霸主地位,很有可能成为史上最畅销的丙肝药物。
前药研究的又一胜利:乙肝新药Vemlidy
2016年11月、12月,吉利德开发的新药Vemlidy(TAF,替诺福韦艾拉酚胺富马酸)先后获得美国FDA、日本厚生劳动省的批准,用于慢性乙型肝炎成人感染者的治疗。同时,欧洲药品管理局人用医药产品委员会(CHMP)也表示支持Vemlidy 用于慢性乙型肝炎成人感染者以及年龄≥12岁且体重≥35公斤的青少年感染者的治疗,在2017年第一季度有望获批在欧盟上市。
据估计,在全球范围内有多达3.5亿~4亿乙肝患者,该病可导致肝硬化,是全球80%原发性肝癌的直接病因。中国是乙肝大国,据保守估计,全国13亿人口中有1亿慢性乙型肝炎病毒感染者,约占全球乙肝携带者的1/3,而且中国乙肝发病率还在持续上升。目前全球最畅销的乙肝药物是恩替卡韦,Vemlidy的获批,必将改变乙肝药物市场的格局。
Vemlidy(图3)是一种新型核苷类逆转录酶*制剂抑**(NRTI),该药是吉利德已上市药物Viread(替诺福韦酯,TDF)的前药。因为TAF 具有较高的血液稳定性,可以有效递达至肝细胞,因此TAF 在剂量低于Viread 十分之一(25 mg/300 mg)的情况下就能发挥与后者相似的疗效,同时还可避免血液中替诺福韦浓度过高,提高了安全性。
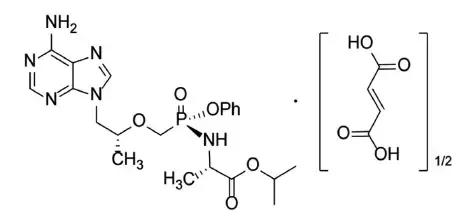
图3 Vemlidy 结构式
不过戏剧性的是,早在2001年,吉利德的科学家在一项动物研究上,已经发现TAF的抗HIV活性是替诺福韦的1000倍以上,且TAF在毒性方面具有更大的优势。尽管获得了上述积极结果,吉利德还是在2004 年突然宣布中止TAF的研究项目。直到2010年,吉利德才再次开始公开大力宣传之前TAF的研究结果。Vemlidy的推迟上市,也使得Viread在2018年专利保护到期前多卖了数十亿美元,这也招致了患者及护理人员的谴责,甚至是法律诉讼,指控吉利德为了攫取更多利润而故意推迟安全性更高的新药的上市时间。
基于TAF 的专利布局:Genvoya、Odetsey 和Descovy
替诺福韦酯在20世纪80年代被欧洲的科学家发现,当时还是小型生物科技公司的吉利德买下了替诺福韦酯的开发权利并在之后发现其具有抗HIV病毒的作用。吉利德之后基于替诺福韦先后推出了多种每日口服1次的多合一复合片剂(Truvada、Atripla、Stribild、Complera),逐步统治了HIV药物市场。到2009年,吉利德的市值超过了另一制药巨头礼来,其70亿美元销售收入中80%都来自于包括替诺福韦酯在内的艾滋病药物。
而吉利德并没有就此止步,继续研发替诺福韦酯前药,提高疗效和安全性。不仅在2016年以Vemlidy 的商品名上市治疗乙肝,还将TAF 与其他抗病毒药物、药物增效剂制成固定复方口服片剂,用于HIV的治疗,仅半年时间,就上市了3款药物。
2015年11月,以TAF为基础的第一个治疗药物Genvoya 被美国和欧盟批准作为治疗12岁以上的儿童和成人HIV-1感染的指定用药。Genvoya是一种包含elvitegravir(埃替拉韦)、emtricitabine(恩曲他滨)、药物增效剂cobicistat及TAF组成的复合药物。
2016 年3 月,FDA 批准吉利的Odefsey 治疗HIV- 1 感染的患者。Odefsey 是吉利德获得的第2 个关于TAF的固定复方口服片剂,也是单一片剂治疗HIV的最小片剂的代表。其组分为恩曲他滨、rilpivirine(利匹韦林)和TAF这3种。
2016 年4 月,Descovy 成为第3 个以TAF为基础的治疗药。Descovy的组分为恩曲他滨和TAF两种。
这3个药品虽然都不能治愈HIV感染,只能尽最大程度改善,患者的生存质量。但能被欧美药监系统批准上市3个TAF组合制剂,可见吉利德在抗病毒药物专利布局、临床试验及政府公关上的不凡手段。
靶向抗肿瘤用药任重道远
随着人们对细胞周期及癌症细胞特异性的深入研究,人类对抗癌症的手段不断进步,从放疗到第一个化疗药物*芥氮**,从靶向治疗药物的鼻祖Rituxan到第一个宫颈癌疫苗Gardasil的获批。抗肿瘤药物的研发日新月异,也给制药企业创造了巨大的商机。近5年来,靶向抗癌新药的捷报频传。2016年,美国FDA批准了4个靶向抗癌新药。
首个蛋白相互作用(PPI)*制剂抑**:Venclexta
2016 年4 月,由艾伯维公司(AbbVie)与罗氏制药(Roche)合作开发的一款突破性抗癌药Venclexta(图4)获美国FDA批准上市,用于治疗携带17p删除突变(del 17p)以及之前已接受至少一种疗法的慢性淋巴细胞白血病患者。

图4 Venclexta 结构式
Venetoclax是口服小分子B细胞淋巴瘤因子-2(BCL-2)*制剂抑**。在慢性淋巴细胞白血病肿瘤细胞中,BCL-2的过度表达已被证明介导肿瘤细胞抗凋亡,并与化疗耐受性有关。Venetoclax通过直接与BCL-2 蛋白结合,代替促凋亡蛋白质引发线粒体外膜透化,进而启动肿瘤细胞凋亡。
蛋白-蛋白相互作用*制剂抑**(PPI)靶点不同于传统的药物靶点,其结合位点位于蛋白质表面,面积非常大(1000~6000Å2),但口袋非常浅,想通过一个小分子阻断两个蛋白质的结合非常不易,所以要求配体比传统药物分子更大。这也导致PPI小分子*制剂抑**会面临溶解性、细胞渗透性、口服生物利用度等诸多药代动力学问题,成药性显著下降。因此,PPI靶点药物能成功进入临床II 期并保持活性的非常少。Venetoclax以每日一次口服,单独用药治疗慢性淋巴细胞白血病获批,给制药界打了一剂强心剂。
首个PD-L1 抗体药物:Tecentriq
2016年5月,罗氏在研PD-L1免疫疗法药物Tecentriq 在美国提前4个月获得FDA 加速批准,用于治疗最常见类型的膀胱癌——尿路上皮癌,该药是FDA 批准的首个PD-L1 免疫疗法药物,同时也是获批治疗膀胱癌的首个PD-1/PD-L1免疫疗法药物。
膀胱癌是全球第9大最常见癌症,男性发病率为女性3倍。转移性尿路上皮膀胱癌治疗选择有限,而且预后差,在近30 年中该领域无重大进展。Tecentriq的上市,将为转移性尿路上皮癌群体提供一种重要的治疗选择。2016 年10 月,FDA 再次批准Tecentriq用于接受含铂化疗治疗期间或治疗后病情进展及接受靶向疗法(若肿瘤中存在EGFR或ALK基因异常)治疗失败的转移性非小细胞肺癌患者。
有消息显示,罗氏正在积极推进一个庞大的临床开发项目,调查Tecentriq治疗特定类型肺癌、肾癌、乳腺癌和膀胱癌的潜力。同时,罗氏也正在努力推进Tecentriq 与其他药物的组合疗法,以挖掘该药的最大临床潜力。
成人软组织肉瘤患者的福音:Lartruvo
2016年10月、11月,礼来公司(EliLilly)开发的新药Lartruvo先后获得美国FDA、欧盟委员会的批准。Lartruvo与阿霉素(doxorubicin)联用,用于组织学亚型为适合含蒽环类方案并且不适合采用放疗或手术根治的软组织肉瘤成人患者。
软组织肉瘤是对发生于全身软组织(脂肪、肌肉、神经、纤维组织、血管)的一大类恶性肿瘤的统称,非常复杂并具有多种亚型,使得难以诊断和治疗。近几十年来,能够延长晚期软组织肉瘤总生存期的一线治疗方面始终没有进展。美国癌症研究协会的数据显示,2015年,在美国确诊1.2万软组织肉瘤新病例,约5000例死亡病例。
此次批准,使Lartruvo与阿霉素的联合用药方案成为过去40年以来FDA批准用于软组织肉瘤的首个一线疗法,标志着在晚期软组织肉瘤临床治疗迈出重要一步。临床试验数据显示,与阿霉素单药治疗相比,Lartruvo联合阿霉素使患者总生存期提高11.8个月。
Lartruvo是一种人血小板衍生生长因子受体α(PDGFRα)拮抗剂,该药是FDA批准治疗软组织肉瘤的首个单克隆抗体药物。当PDGF 受体被相关配体刺激之后,下游信号通路可引起肿瘤生长。Lartruvo通过阻断这些受体来有效减缓或终止肿瘤生长(图5)。
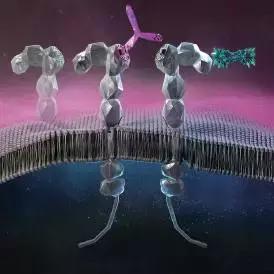
图5 Lartruvo(红色)与PDGFR-α受体(灰色)结合,阻断PDGFR-α信号通路示意
中枢神经系统用药表现不俗
由于中枢神经系统疾病的特殊性,中枢神经系统新药的发现往往出自临床的偶然现象。科研工作者在研发中枢神经系统用药的同时,也在不断探索这类疾病的新机理。这也就伴随着新药物的研发。据统计,中枢神经系统用药的新药的II、III期临床试验通过率为9% ,低于所有药物的平均通过率11%。2016年,美国FDA一连批准了4个中枢神经系统用药,可谓是这一领域的突破之年。
抗癫痫药物新秀:布瓦西坦(Briviact)
2016 年1 月、2 月,比利时优时比(UCB)公司的布瓦西坦(图6)分别通过欧洲委员会和美国FDA 的批准上市,用于癫痫患者的部分发作,伴有或不伴继发性全身性发作的辅助治疗。
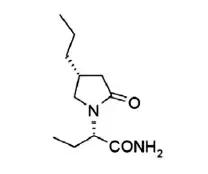
图6 布瓦西坦结构式
癫痫是一种长期、慢性脑部疾病,以脑内神经元群反复发作性过度放电而引起的突发性、暂时性脑功能失常为特征的经系统常见病。癫痫在人群中的发病率为0.6%~1.1%,其中儿童癫痫发病率约为成人10~15倍,约60%癫痫患者起病于儿童期。目前全球约有5000万癫痫患者,每年新增200万癫痫患者。中国约有1000万癫痫患者,其中600多万为有发作的患者,每年新发癫痫患40万人。
布瓦西坦是UCB 公司的开浦兰(左乙拉西坦)遭遇专利悬崖后,推出的一款新型的抗癫痫药物。布瓦西坦通过与突触囊泡蛋白2A(SV2A)结合而发挥抗癫痫作用。相较左乙拉西坦,布瓦西坦与SV2A 的结合力约为左拉西坦的10倍,且生物利用度高,达峰时间短。反复给药后1周内达到稳定血药浓度,且食物对其吸收无影响。布瓦西坦良好的药理学活性、临床疗效及安全性有望成为继左乙拉西坦后又一重磅抗癫痫药物。
变废为宝的Zinbryta
2016 年5 月,美国FDA 批准了百健艾迪(Biogen Idec)和艾伯维合作开发的Zinbryta(达利珠单抗)用于复发性多发性硬化症成年患者治疗。
实际上,达利珠单抗并非一个新药,1997 年罗氏开发的达利珠单抗曾以商品名Zenapax上市,用于器官移植的免疫抑制。后因销量不佳,Zenapax在2009年退市。百健在前者基础上做了工艺改造,得到了糖基化修饰一致性更好、抗体依赖的细胞介导的细胞毒性作用(ADCC)活性更低的高产达利珠单抗,半衰期延长约30%,并针对新适应症进行临床研究。
III期临床试验数据显示,与每周1次肌肉注射药物Avonex(干扰素β-1a)相比,每月皮下注射1次的Zinbryta在减少疾病活动方面表现出了统计学意义的显著改善,包括降低年度复发率和新的脑部损伤的形成。而Avonex是百健艾迪最畅销的多发性硬化症药物,位列多发性硬化症药物市场全球第二。Zinbryta的上市,将巩固百健艾迪在多发性硬化症市场的地位。
唯一获批的脊髓性肌萎缩症治疗药物:Spinraza
2016年12月,由百健与Ionis制药公司合作开发的一款罕见病治疗药物Spinraza获FDA批准,用于脊髓性肌萎缩症儿科患者和成人患者的治疗。此次批准,使Spinraza成为全球首个也是唯一一个获批治疗脊髓性肌萎缩症的药物。
Spinraza 是一种反义寡核苷酸(ASO),旨在改变SMN2 基因的剪接,以增加全功能性SMN蛋白的生产;在临床研究中,Spinraza治疗显著提高了脊髓性肌萎缩(SMA)患者的运动机能。
值得一提的是,FDA 自收到Spinraza监管文件之后的3个月内便加速批准Spinraza,突显了该领域迫切的严重未满足的巨大医疗需求。在美国和欧盟,Spinraza均被授予孤儿药地位;在美国,Spinraza还获得了快车道地位和优先审查资格;在欧盟也收获了加速审批资格。
脊髓性肌萎缩症是一种会导致肌肉无力和萎缩的运动神经元性疾病,该病属于基因缺陷导致的常染色体隐性遗传病,对患者周身上下的肌肉都会造成侵害,患者主要表现为全身肌肉萎缩无力,身体逐渐丧失各种运动功能,甚至是呼吸和吞咽。脊髓性肌萎缩症是2岁以下婴幼儿群体中的头号遗传病杀手,该病是一种相对常见的“罕见病”,在新生儿中的患病率为1∶6000~1∶10000。
曲折中前行的生物类似药
小分子药物的专利悬崖还未走远,生物药专利在2014—2020年也迎来了集中到期时间段。IMS 预测,至2020年,生物类似药的年销售额有望达到250 亿美元,约占生物药市场份额的10%。自从2013年EMA批准第一个单抗类似药Inflectra(参照药为英利昔单抗)开始,全球的生物类似药的热潮迅速燃起。2015年3月,FDA也批准了美国第一个生物类似药Zarzio(参照药为非格司亭)上市。
2016 年,Celltrion 的Inflectra 终于登陆美国市场。此外,山德士公司的Erelzi(参照药为依那西普)和安进制药的Amjevita(参照药为阿达木单抗)也分别在2016年8月和9月获批。但是2016 年对于生物类似药并非一帆风顺,安进和山德士还在就Erelzi何时上市在法庭争论不休。安进和艾伯维的官司也在进行中,Amjevita 很可能在2018年之前都无法上市。对于患者来讲,这也可以说是美国药物审批和专利诉讼双线进行的弊端。
国内药物领域重要事件
2016年,国内药物相关新闻频现,周德敏/张礼和研究团队在病毒疫苗领域取得重大突破,仿制药一致性评价政策陆续出台,《中华人民共和国中医药法》颁布,给国内医药行业的研发、生产、监管带来一系列重要的影响。
周德敏/张礼和研究团队在病毒疫苗领域取得重大突破
2016年12月,周德敏/张礼和研究团队的一篇研究报告在《Science》发表,引起了海内外的轰动。该团队研制出了具有完整病毒结构,可快速制备的活流感病毒疫苗。更令人兴奋的是,这一成果或将普遍适用于几乎所有病毒,有望彻底改变人类对抗病毒的研发思路,成为首个广谱性抗病毒疗法。
目前广泛使用的疫苗主要包括灭活疫苗和减毒活疫苗。灭活疫苗在灭活过程中,病毒的蛋白质结构会发生改变,因此疫苗的免疫原性有限。减毒活疫苗虽然保留了良好的免疫原性,但存在免疫逃逸的可能性。
而周德敏/张礼和研究团队的成果综合了灭活疫苗和减毒活疫苗的优点,他们在(甲型)流感病毒的信使RNA保守区(不易突变位点,一旦突变,病毒即死亡)中引入多个终止密码子,并保留病毒的完整结构。这一修饰既保留了病毒良好的免疫原性,可激活宿主的全部免疫反应。同时由于终止密码子的存在,病毒无法进行蛋白质翻译,进而阻止病毒在宿主体内的复制,不会使宿主致病。
为了制备这些特殊的活病毒疫苗,周德敏/张礼和研究团队还建立了一套专门的活病毒疫苗生产体系,利用非天然氨基酸、与这类氨基酸匹配的转运RNA(tRNA)以及tRNA合成酶组装活病毒疫苗,保证量产(图7)。
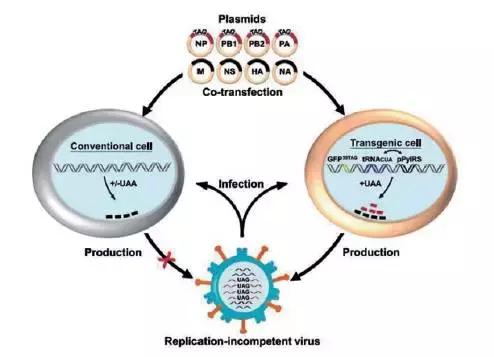
图7 活病毒疫苗组装及感染宿主示意
仿制药一致性评价正式拉开序幕
自从2012年国家食品药品监督管理总局(CFDA)成立仿制药质量一致性评价办公室以来,仿制药一致性评价工作如何展开一直没有明确。直到2016 年3 月5 日,国务院办公厅发布《关于开展仿制药质量和疗效一致性评价的意见》,仿制药一致性评价的大幕才正式拉开。截至2016 年12 月共发布与仿制药一致性评价工作相关的10个工作文件、8个一致性评价征求意见、1个意见、5个公告、1个通告、2个通知、1 个文号信息和5 个指导原则。此外,CFDA专门成立专家委员会,负责审议参比制剂的选择和审议仿制药品种评价结果。
总体来讲,仿制药一致性评价工作,有利于提高国产仿制药竞争力、淘汰落后产能、降低医药总费用支出。可以预见,未来的3~5年内,国内制药行业必将经历翻天覆地的变化。
《中华人民共和国中医药法》出台
2016年12月25日,十二届全国人大常委会第二十五次会议审议通过了《中华人民共和国中医药法》,并将于2017年7月1日正式实施。
中医药是中华民族的瑰宝,是中国独特的卫生资源、潜力巨大的经济资源、具有原创优势的科技资源、优秀的文化资源和重要的生态资源。但是,中医药法制建设相对滞后,对中医药事业健康、持续、稳定发展带来了极大的不确定性因素。《中华人民共和国中医药法》作为第一部全面、系统体现中医药特点的综合性法律,对于中医药行业发展具有里程碑意义。(责任编辑 田恬)
作者简介 :李辉,中国医药保健品进出口商会,研究员,研究方向为医药外贸、医药国际政策。
注 :本文发表在2017年第1期《科技导报》,欢迎关注。