
天然产物、复杂的靶点、先导化合物的优化,将这些梳理为PAR-1及其拮抗剂的关键词,绝不为过。结构复杂的天然产物经过化学合成方法的精雕细琢并最终成药,这不仅展示了当前高超的全合成技术,同时,还拉开了PAR-1拮抗剂的发展序幕。不过,虽历经多年的探索,PAR-1已有相应药物上市,但何时能跨入抗血小板第一梯队,一直饱受猜疑。下面,小编将对药物靶点PAR-1及其拮抗剂的发现过程进行简要梳理,如能对君有所帮助,那定是再好不过的事了。
1药物靶点的发现
众所周知,凝血酶可以激活血小板且活性非常强,但作为一个丝氨酸蛋白酶大分子,它是如何激活血小板聚集的机制却一直不清楚。直到1991年,美国加州大学旧金山分校Coughlin教授团队克隆了人体血小板凝血酶受体,发现凝血酶是通过凝血酶受体激活血小板聚集的,而且是一种新型的激活机制,见图一。
这种机制的具体过程是:凝血酶与血小板表面的凝血酶受体氮端结合并将其水解切断,产生出新的氮端,新的氮端通过分子内反应与凝血酶受体自身结合,从而激活凝血酶受体,引发细胞内信号的传递,最终导致血小板聚集。所以,凝血酶受体也称为蛋白酶激活受体-1(PAR-1)。随后,又陆续发现了蛋白酶激活的受体2、3、4亚型。另外,新氮端的前6个氨基酸多肽(丝氨酸-苯丙氨酸-亮氨酸-亮氨酸-精氨酸-天冬酰胺)也可以激活凝血酶受体,被称为凝血酶受体活化肽(TRAP),常用于体外和体内凝血酶受体的活化研究。
在人体内,血小板主要表达为PAR-1与PAR-4,且两者均可被凝血酶激活而诱导血小板分泌与聚集,但凝血酶对PAR-1的亲和力比PAR-4高40倍。PAR-1尚存在于内皮细胞、平滑肌、成纤维细胞及心肌细胞,这类细胞中PAR-1经凝血酶介导而激活后,诱导炎症反应及引发细胞增生。因此,PAR-1拮抗剂不仅可降低动脉血栓的形成,还可调节包括瓣膜术后的再狭窄等其他凝血酶介导的途径。
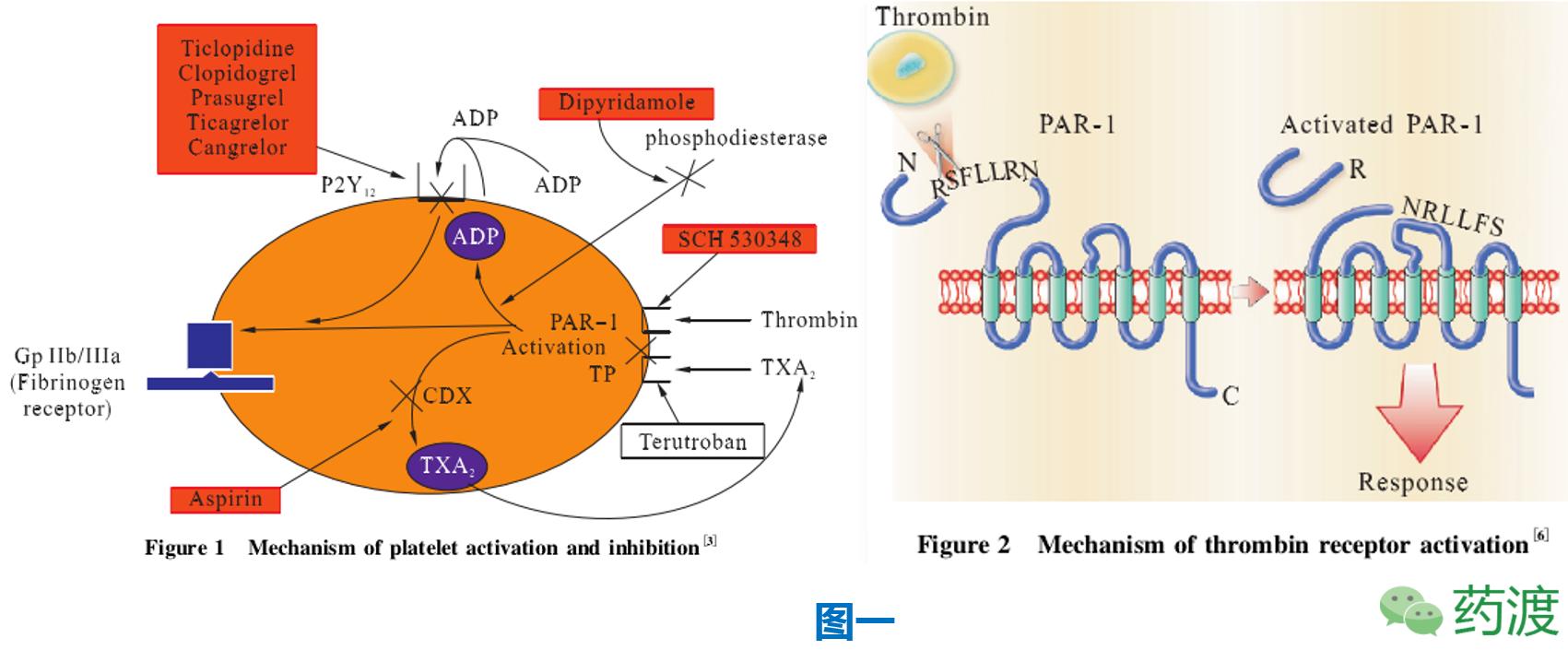
PAR-1拮抗剂、喜巴辛类似物的发展
PAR-1的发现为研发新型抗血栓药物提供了一个新的药物机制和靶点,引起了学术界和制药业的广泛关注。20多年来,全球多个研究组报道了数种不同系列的凝血酶受体拮抗剂,但由于凝血酶受体的特殊活化机制,只有少数化合物被发现具有较好的凝血酶受体拮抗活性。
喜巴辛,作为一种天然产物,最初研究的目的是为了寻找抗阿尔茨海默病药物。研究发现,喜巴辛对乙酰胆碱毒蕈碱受体M2有强效抑制作用,且IC50为4.5nM,选择性强于M1约10倍。为了获得活性更强选择性更高的M2受体拮抗剂,大量的喜巴辛类似物被合成出来,后来,随着研究的深入,喜巴辛及其类似物经随机筛选,成功的转轨至研发PAR-1拮抗剂的道路上。
为了获得活性更好的喜巴辛类化合物,国外如Merck、法国PierreFabre研究中心、德国拜耳、Schering-Plough、韩国化学研究院、LG生命科学等制药公司,以及我国的科研院所和大型制药公司等科研人员对其进行了结构改造及活性筛选,经筛选后的这类化合物对PAR-1的IC50值大体上在10~500nM之间,其中,vorapaxar作为喜巴辛类似物、PAR-1受体拮抗剂脱颖而出,并最终成药。见图二。
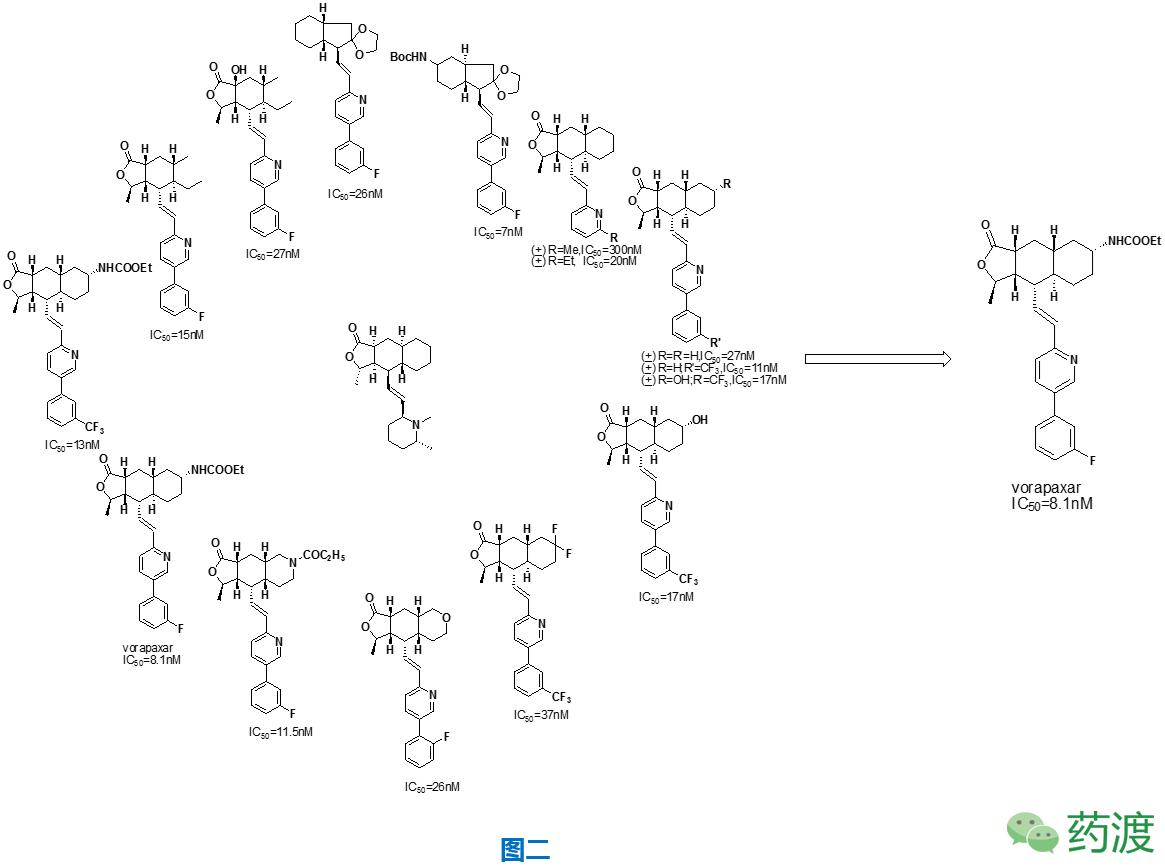
在喜巴新类似物vorapaxar循序渐进的研发过程中,另一类作用于PAR-1的拮抗剂也在如火如荼的跟进,他们就是异吲哚啉类化合物(见图三),其中,各方面相对表现优秀的是日本卫材的atopaxar。atopaxar (E5555)作为一种口服强效、具有选择性的PAR-1受体拮抗剂,口服3.5h即作用达峰,后通过肝脏CYP3A4酶代谢,经粪便排泄,半衰期为23h。但该候选化合物做到临床二期,便终止了开发,具体原因未对外公开。除了异吲哚啉类化合物对PAR-1存在较好的拮抗作用,如图四所示的一些结构也表现了较好的效果。
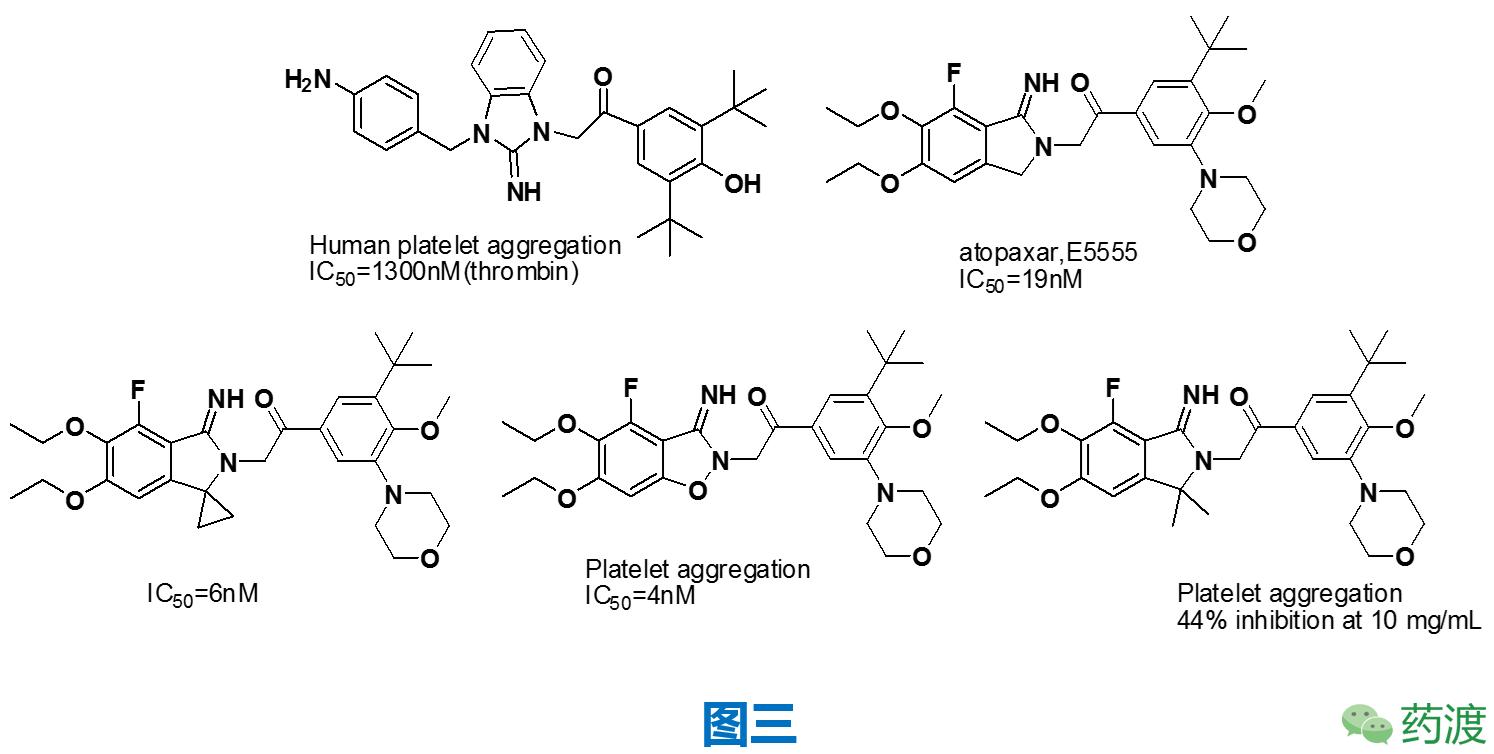
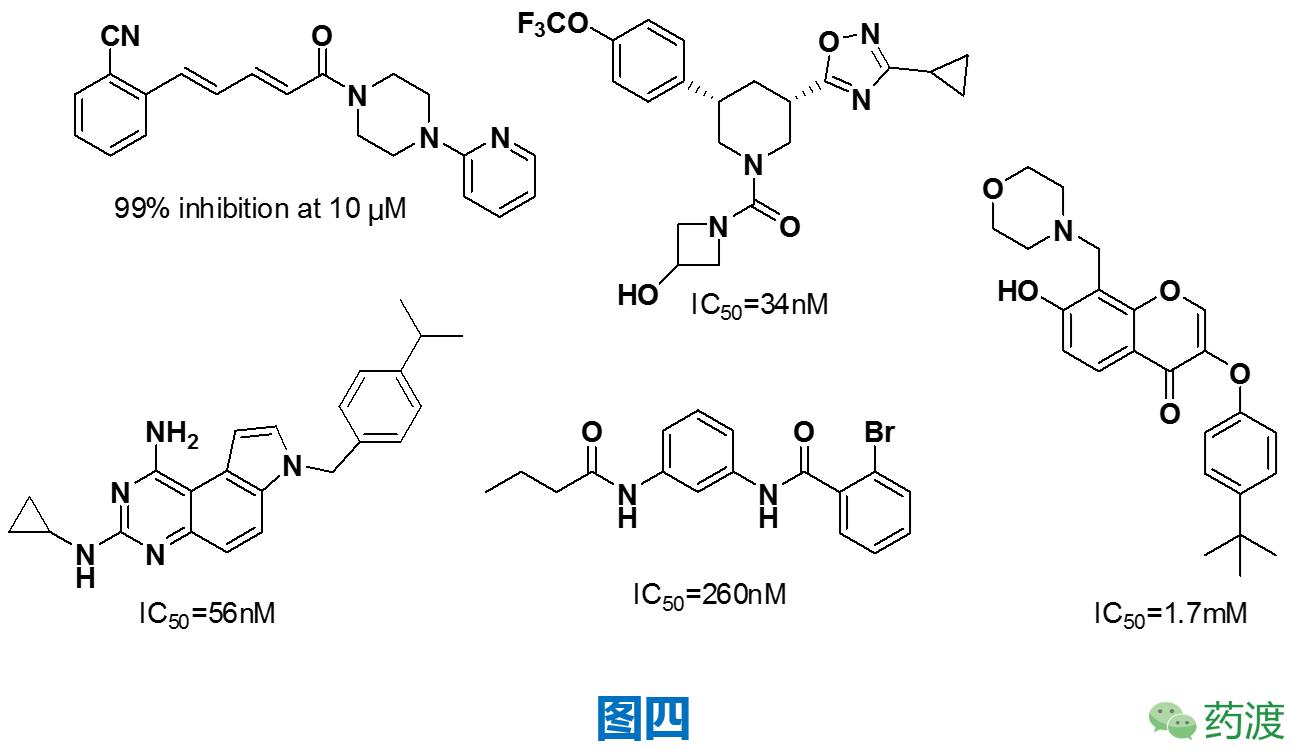
硫酸沃拉帕沙的上市之路
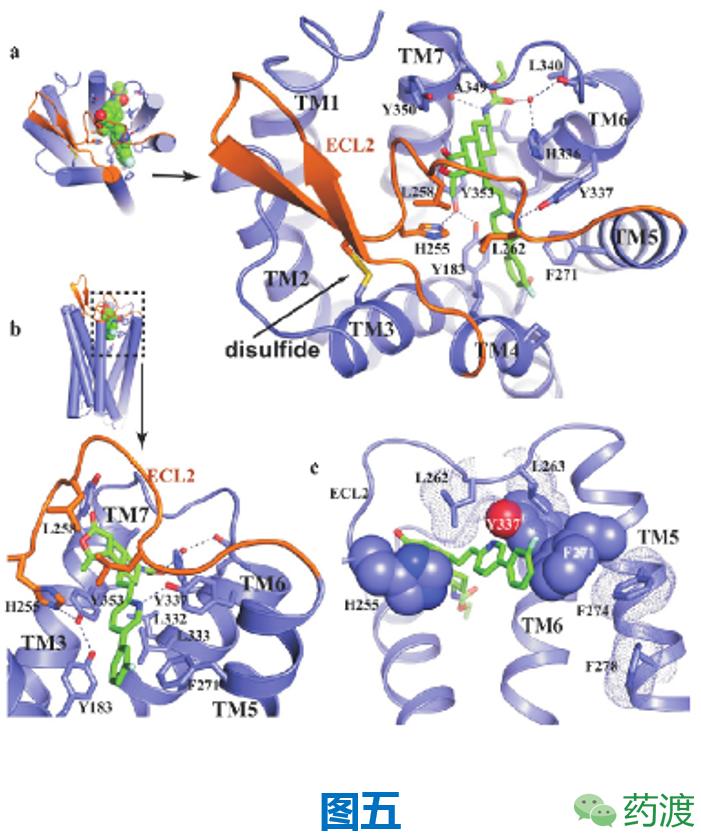
早年间,Coughlin教授与斯坦福大学的kobika等便在Nature杂志上发表了关于vorapaxar与人体PAR-1结合的高分辨晶体结构(见图五),这个结构显示,vorapaxar与PAR-1的结合区域是在PAR-1表面比较浅的地方,而且很少与周围的水环境有接触,这是vorapaxar与其他具有较深并接触水环境结合区域的G蛋白偶联受体不同的地方。另外,PAR-1的Tyr337与vorapaxar的吡啶环形成强的氢键、Phe271与vorapaxar的氟代苯环也互相存在作用,这些可能是vorapaxar对PAR-1有亲和性和选择性的原因之一。
经过多年的努力,2013年7月24日,Merck公司宣布,美国食品与药品管理局(FDA) 接受了将vorapaxar用于对有心脏病发作史但没有短暂性脑缺血发作史或中风史的患者进行心血管病预防的新药申请(NDA)。2014年5月8日,vorapaxar (商品名:Zontivity)终获FDA批准上市,临床上用于遭受心脏病发作的患者或腿部动脉有堵塞的患者,以降低进一步的心脏病发作、中风、心血管死亡和需要手术的风险。作为一种首创(first-in-class)的PAR-1拮抗剂,也是一种抗血小板*制剂抑**,旨在减少血小板聚集,抑制血块的形成,适用为心肌梗死(MI)或有外周动脉疾病(PAD)史患者中的血栓性心血管疾病,可显著减低心血管栓塞、MI、中风和急性冠状动脉血栓发生率。
虽然,Zontivity成功上市,但其后续的发展似乎并不平坦。业界曾经对其预测,峰值销售为50亿,但后来因为临床试验数据不佳,业界估计只有3亿美元的峰值销售。据汤森路透数据显示,2015年,Zontivity的全年销售额仅有2000万美元,并没有展现出强劲的势头。且在销售额不尽人意的状况下,2016年8月,默沙东也对外宣布,将停止在美国继续推广Zontivity,并因此裁减148名市场销售员工。故Zontivity是否会成为下一个“死药”,业界对此深表怀疑。
4未来,路在何方?
研究发现之初,PAR-1拮抗剂一直被期待成为不增加出血风险且预防动脉血栓事件的药物。但从临床研究发现,它同样增加出血的风险,尤其增加了卒中史患者颅内出血的风险,除此之外,血管事件总的发生率与对照组并没有明显的区别,不过,心血管事件的发生率倒是确有降低。
经过了这些年的研发,总体看来,PAR-1*制剂抑**还是得到了长足的发展。不得不说,硫酸沃拉帕沙的成功上市,吸引了更多的制药公司来开发新一代、性能更优越的凝血酶受体拮抗剂,尤其是参照vorapaxar-PAR-1晶体结构进行药物设计,这让凝血酶受体拮抗剂的研究不在局限于学术界,研发范围更加广阔。
不过,在美国的停止推广,又将vorapaxar蒙上了一层厚重的阴影,同时,也严重的降低了该类药物的研发积极性,尤其是沃拉帕沙类似物的研究。在抗血小板药物这个大市场环境中,面对阿司匹林、氯吡格雷的根深蒂固,PAR-1拮抗剂本就力不从心,再加上上市后如此“坎坷”,PAR-1拮抗剂若想真正的“赶超”前面两位“老大哥”,完成逆袭,似乎还有很长的路要走!
参考文献:
1. 郭宗儒. 基于天然产物首创的凝血酶受体拮抗剂沃拉帕沙[J].药学学报,2016,51(3):496-498。
2. 史建硕,谭伟强,崔 欢等. 凝血酶受体拮抗剂在抗血栓方面的研究进展[J]. 中国药物化学杂志,2014,24(2),129.
3. Yan X, Samuel C, Martin C, et al. Himbacine derived thrombinreceptor(PAR-1) antagonists:SAR of the pyridine ring[J]. Bioorganic &Medicinal Chemistry Letters,2007,17, 4509.
4. Samuel C, Yu GW,William J, et al.Discovery of a Novel, OrallyActiveHimbacine-Based Thrombin ReceptorAntagonist (SCH 530348) withPotentAntiplatelet Activity[J].J. Med. Chem, 2008, 51, 3061.
5. 部分数据来源于汤森路透。
作者信息 :强森
职业从事新药开发
转载声明: 本文为投稿文章,如需转载,必须保留作者信息及本文来源。