Bardoxolone:治疗Alport综合征(遗传性肾炎)
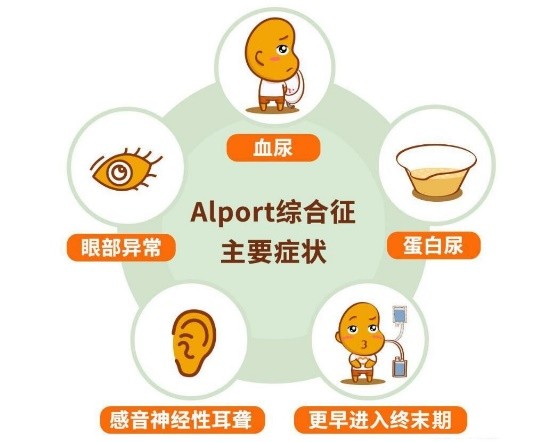
Alport综合征是一种罕见的CKD遗传形式,由编码IV型胶原的基因突变引起,IV型胶原是肾脏肾小球基底膜的主要结构成分。Alport综合征影响儿童和成人。Alport综合征患者的肾脏逐渐失去从血液中过滤废物的能力,这可导致终末期肾病,需要进行慢性透析治疗或肾移植。在病情最严重的患者中,大约50%在25岁时进展到透析,90%在40岁时进展到透析,近100%在60岁时都会进展到透析。根据Alport综合症基金会,Alport综合症影响美国大约30000到60000人。目前还没有批准治疗Alport综合征引起的CKD的疗法。
Reata Pharmaceuticals是一家处于临床阶段的生物制药公司,该公司前不久宣布向欧洲药品管理局(EMA)提交Bardoxolone methyl (“Bardoxolone”)的上市许可申请(MAA),用于治疗Alport综合征(Alport syndrome)引起的慢性肾病(CKD)。
Bardoxolone是一种每天口服一次的研究性Nrf2激活剂,Nrf2是一种转录因子,通过恢复线粒体功能、减少氧化应激和抑制促炎症信号传导诱导促进炎症消退的分子途径。FDA已授予bardoxolone孤儿药资格认定,用于治疗Alport综合征和常染色体显性多囊肾病(“ADPKD”)。欧盟委员会也已授予bardoxolone治疗Alport综合征的孤儿药资格认定。
MAA提交基于CARDINAL 3期临床试验的疗效和安全性数据。在此之前,美国食品和药物管理局(“FDA”)已受理该公司的新药申请(“NDA”),用于治疗Alport综合征引起的慢性肾病,并将在2022年2月25日前给出最终决定。
除了治疗Alport综合征引起的CKD的CARDINAL 3期研究外,Bardoxolone目前还正在FALCON中进行研究,用于治疗ADPKD引起的CKD的3期研究;MERLIN,治疗有快速进展风险的CKD患者的2期研究和AYAME,同时RETA的许可方Kyowa Kirin正在日本进行糖尿病肾病治疗的3期研究。Bardoxolone治疗对由ADPKD、IgA肾病、局灶性节段性肾小球硬化和1型糖尿病引起的CKD患者的2期研究产生了积极的结果。
Kyowa Kirin也在日本向卫生、劳动和福利部提交了一份关于Bardoxolone改善Alport综合征患者肾功能的NDA,该申请目前正在审评中。
Eplontersen:治疗转甲状腺素蛋白淀粉样变性(ATTR)新药
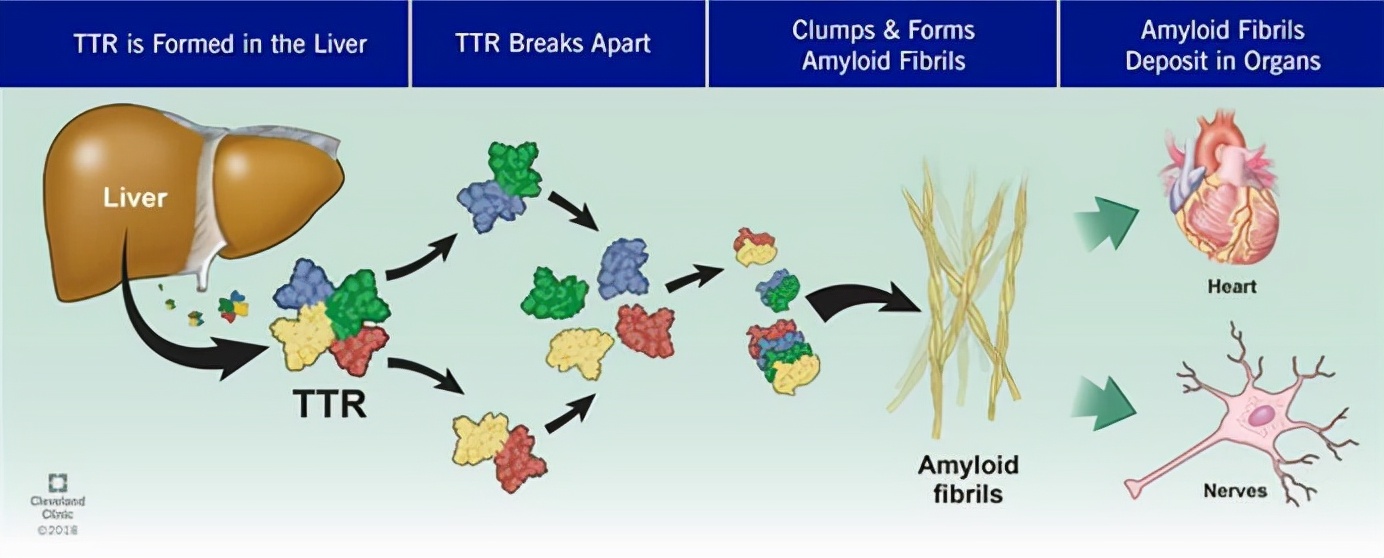
ATTR淀粉样变性是一种罕见的系统性、进行性、致死性疾病,其特征是:异常解离错误折叠的转甲状腺素蛋白(TTR)构成的淀粉样沉积物,在人体各种器官和组织中的异常形成和沉积,包括周围神经、心脏、肠道、眼睛、肾脏、中枢神经系统、甲状腺、骨髓等。TTR淀粉样沉积物在这些组织和器官中的逐渐累积,可导致器官衰竭,最终导致死亡。
转甲状腺素蛋白淀粉样变性心肌病(ATTR-CM)和转甲状腺素蛋白淀粉样变性多发性神经病(ATTR-PN)是ATTR淀粉样变性的2种临床表现形式。ATTR-CM是一种系统性、渐进性、致命性疾病,可在确诊后4年内导致进行性心力衰竭和死亡。由于缺乏对疾病的认识以及症状的异质性,该病仍然未得到充分诊断,其发病率被认为被低估。遗传性ATTR-PN是一种使人衰弱的疾病,在诊断后5年内导致周围神经损伤和运动障碍,如果不治疗,通常在10年内死亡。
2021年12月,阿斯利康(AstraZeneca)与Ionis达成战略合作,开发和商业化Eplontersen。近日,Ionis Pharma宣布,美国食品和药物管理局(FDA)已授予Eplontersen孤儿药资格ODD(孤儿药(Orphan Drug)是指用于预防、治疗、诊断罕见病的药品,而罕见病是一类发病率极低的疾病的总称,又称“孤儿病”。在美国,罕见病是指患病人群少于20万的疾病类型,罕见病药物研发方面的激励措施包括各种临床开发激励措施,如临床试验费用相关的税收抵免、FDA用户费减免、临床试验设计中FDA的协助,以及药物上市后针对所批准适应症为期7年的市场独占期)。
Eplontersen是一种配体偶联反义(LICA)药物,可减少转甲状腺素蛋白(TTR)的产生,用于治疗遗传型和非遗传型TTR淀粉样变性(ATTR)。目前,Eplontersen正在3期临床试验中测试,用于治疗转甲状腺素蛋白淀粉样变性心肌病(ATTR-CM)和转甲状腺素蛋白淀粉样变性多发性神经病(ATTR-PN)。
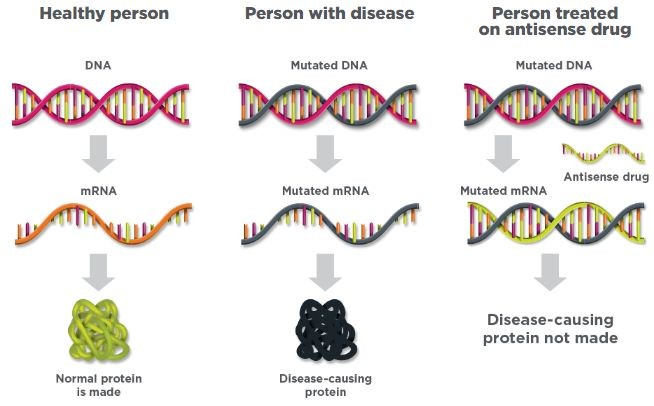
反义药物作用机制(图片来源:Ionis Pharma)
NR082:引领中国眼科体内基因治疗走进了临床时代
2022年1月18日,中国眼科疾病的体内基因治疗领导者纽福斯生物科技有限公司(下称“纽福斯”)宣布公司的候选药物NR082(rAAV2-ND4,核心项目NFS-01),用于治疗ND4突变引起的Leber遗传性视神经病变(ND4-LHON)获美国食品药品监督管理局(FDA)授予新药临床试验(IND)许可,并将在美国开展临床试验。NR082是首个获得美国FDA临床试验许可的中国籍眼科体内基因治疗药物。
Leber遗传性视神经病变典型的首发症状是视物模糊,随后的几个月之内出现无痛性、完全或接近完全的失明,通常是两眼都受累,或者一只眼睛失明不久,另一只也很快失明。视神经和视网膜神经元的退化是LHON的主要病理特性。另外还有周围神经退化、震颤、心脏传导阻滞和肌张力的降低。LHON通常在20~30岁时发病,但发病年龄范围可从儿童时期到70多岁。该病通常存在性别差异,男性患病风险一般是女性的4~5倍,但原因尚不清楚。突变基因位于线粒体基因组,而胚胎的线粒体都来自卵子,因此该突变仅由母系遗传,男性并不会把致病基因传给子代。

NR082是一款处于研发阶段的体内基因治疗产品,此前已获得美国FDA孤儿药认定并引领中国眼科体内基因治疗走进了临床时代。此次在美国获批开展的临床试验是一项多中心、开放的、单臂临床研究,该项研究将在ND4线粒体基因突变引起的Leber遗传性视神经病变(LHON)患者中评价NR082基因治疗的安全性和有效性。
NR082是第一个获得FDA授予IND许可的中国籍AAV体内基因治疗药物,这是中国开发的基因治疗药物能在眼科领域为患者带来光明希望的重要里程碑,也为实现眼科患者带去革命性基因治疗手段的目标更进一步。
参考资料:
1. Reata Pharmaceuticals Submits Marketing Authorization Application to the European Medicines Agency for Bardoxolone Methyl in Chronic Kidney Disease Caused by Alport Syndrome
2. Ionis announces eplontersen receives orphan drug designation from U.S. FDA
3. Anthony K M , Patrick Y , Alex K , et al. Gene–environment interactions in Leber hereditary optic neuropathy[J]. Brain, 2009, 132(Pt 9):2317-2326.