Gaetano Thiene

图1. Gaetano Thiene(生于1947年7月1日)是意大利帕多瓦大学心血管病理学名誉教授。他的专业兴趣包括心脏病学和病理学。Thiene于1972年获得医学学位,并于1975年至1978年期间从事心脏病学和病理学专业的研究生工作。他随后的专业职位包括病理学教授、心血管科学和病理解剖学研究所所长、医学诊断科学和特殊疗法系副主任(均在帕多瓦大学),并于2002年成为帕多瓦大学荣誉院士皇家医师学院。
世界儿科和先天性心脏手术学会一直邀请心态学家发表“Stella van Praagh”演讲,我(Gaetano Thiene)感到非常荣幸能够出现在这个名人堂上。Dick Van Praagh和Bob Anderson都是我以前的导师,他们启发了我对先天性心脏病(CHD)研究的兴趣。如果我在早期的职业生涯中能够望向这个研究领域的地平线,那是因为我站在巨人的肩膀上。
作为心脏病学家和病理学家,我的大部分职业生涯都致力于病理生理学,这与我的大学的悠久传统相符,那里的临床病理学相关方法是由Giovanni Battista Morgagni(1682-1771)引入的。通过对同一位患者在死亡前和死亡后进行尸检,他能够通过病变器官的重构找到体征和症状的病理生理学解释。他预见了转化医学的概念,这就是为什么我们将“G. B. Morgagni”命名为我们的“专科转化医学”博士课程的原因。
我选择艾森曼格综合症作为我的演讲主题,并不是偶然的,因为它是心血管病理生理学的典范。我只是想提醒读者,正是在帕多瓦大学,Realdo Colombo通过解剖实验发现了小的血液循环,即通过打开活体犬的肺静脉观察到血液而不是空气正在流出,这与Galeno的观点不同。
艾森曼格综合症不仅属于医学史,它仍然作为左右分流性先天性心脏病自然史的严重并发症发生。由于手术延迟,早期发病的意识对改变先天性心脏病的治疗策略起着至关重要的作用,通过在早期婴儿期进行修复手术。不幸的是,特别是在发展中国家,儿科心脏手术几乎不存在,艾森曼格综合症的病例仍然存在。它是成年人中最常见的先天性心脏病之一,也是早逝的原因之一。
维克多-艾森曼格(人物传记)
维克多·艾森曼格(Victor Eisenmenger,1864-1932)出生在奥地利的维也纳,他的父亲奥古斯特·艾森曼格(August Eisenmenger,1830-1907)是维也纳美术学院的教授,是一位著名的肖像和历史题材画家。维克多与安娜·霍伯格(Anna Hoberg,1874-1944)结婚,育有两个女儿:安娜成为了一位才华横溢的雕塑家,而希尔德则成为了一名成功的网球选手。
维克多在维也纳继续学习,在一所奥地利最古老的中学——维也纳学院读书。
1881年至1882年学年,他开始在维也纳大学医学院学习医学,并于1889年2月23日获得医学学位。在1889年至1890年期间,他在维也纳综合医院第二医学科担任助理医师。在此之后的1890年至1893年期间,他担任捷克外科医生、骨科手术创新者爱德华·阿尔伯特(Eduard Albert,1841-1900)教授的第一外科诊所的外科助手。最后,在1894年至1895年期间,他被任命为维也纳第三医学诊所主任、著名的内科医生和喉科医生莱奥波德·舒特(Leopold Schrötter,1837-1908)教授的助手。艾森曼格成为他最喜欢的学生之一,但由于他身体状况不佳,舒特建议他担任哈布斯堡王室的御医,并成为弗朗西斯·费迪南德大公(1863-1914)的私人医生。1895年,艾森曼格被任命为“Titular Hofarzt”(名义上的御医),两年后晋升为“Wirklicher Hofarzt”(实际的御医)。

图 2. A,Victor Eisenmenger(1864-1932)的照片。B,维克多·艾森曼格原始病例报告中的绘图。注意室间隔缺损和指示左向右分流的箭头。
1893年至1902年间,艾森曼格在医学期刊上发表了八篇论文,涉及口腔和咽部肿瘤、假性白血病和心血管疾病,后者包括最终导致艾森曼格综合征名称起源的报告。1920年代后期,他写了一本书,描述了他从1895年到1914年期间作为弗朗西斯·斐迪南大公的私人医生的观察、旅行和经历,当时大公和他的妻子在萨拉热窝被*杀暗**。维克多·艾森曼格于 1932 年 12 月 11 日去世,享年 67 岁。
艾森曼格综合症、综合症和反应
正如Fleming在1997年所强调的那样,Eisenmenger“对这位从幼年时期开始就有青紫症状的患者感到困惑。他无法想象在没有肺动脉狭窄的情况下存在右向左分流,因此认为系统静脉淤血是引起青紫的原因。”“他显然没有意识到肺动脉压力的增加……导致了右向左分流以及青紫。” Rashkind也指出:“Eisenmenger没有讨论肺动脉压力、肺动脉高压或肺动脉病变。他对肺血管方面的全部关注仅限于略微扩张的肺动脉,显示其内表面有内动脉病性增厚,这种病变也延伸到血管的主要分支。”然而,在1898年发表的另一篇文章中,Eisenmenger进一步讨论了主动脉重叠现象,认为它与青紫无关,并假设增加的肺血管阻力可能导致左向右分流的减少,从而确保了青紫的发生。这篇第二篇文章指出了他更为知名的1897年文章的缺陷。
这段话提到了Maude Abbott(1869-1946)在她的《先天性心脏病图谱》一书中引入了“艾森曼格综合征”(就像她为“法洛四联症”一样),意思是“VSD伴随主动脉右位而无肺动脉狭窄或发育不良”。该术语被作者用于表示“艾森曼格”报道的罕见组合,因为没有更好的术语可用。令人惊讶的是,在12年前的文章“先天性心脏病的临床分类及其病理解剖学、诊断和治疗评论”中,Abbott将Eisenmenger的案例归类为“中度发绀”亚型的静脉-动脉分流病例(“morbus caeruleus”),明显认为发绀是由于主动脉从右心室起源,但仍无法解释该综合征的真正病理生理学机制。在缺乏右心导管检查的情况下,正确的解释是不可能的。
确实,转折点发生在1929年,当时 Werner Forssmann (1904-1979) 在他的左臂放置尿路导管并将其插入放射状静脉,成功到达右心室而没有任何心律失常的并发症 (图3)。这标志着右心导管检查的开始。第二天,他的老板解雇了他,并称他这种做法是一种丑闻。Werner Forssmann、André F. Cournand (1895-1988) 和 Dickinson W. Richards (1895-1973) 因他们在心脏导管检查方面的研究成果,于1956年获得了诺贝尔奖。

图 3. Werner Forssmann(1904-1979;A)的照片和胸部 X 光片,输尿管导管插入他的左桡静脉到达右心(B)。
Helen Taussig(1898-1986,图4)在她的书《心脏先天畸形》中报告了一例成年Eisenmenger综合征导致的窒息死亡病例。该患者在波士顿的Peter Brigham医院去世,而Helen正在该院接受培训。尸检后,她在书中报告了该心脏标本的外部和左心室长轴视图(图5)。尸检报告,包括肺的组织学检查,显示“心脏肥大。有大的VSD,主动脉向右偏转并接收来自两个心室的血液。肺动脉异常扩张,并且肺部有显著的改变,类似于动静脉瘤。” 因此,她提出了一种假设,即发绀是由于毛细血管前的动静脉分流引起的,血液绕过了氧合发生的肺泡毛细血管。

图 4. A,海伦·陶西格 (Helen Taussig,1898-1986) 的照片。B,她在 1947 年出版的《先天性心脏畸形》一书的封面。

图 5. 海伦·陶西格 (Helen Taussig) 在她的书中报道的艾森曼格病例。注意扩大的肺动脉、室间隔缺损和主动脉覆盖室间隔。
1947年6月,当她在校正该书的校样时,她加了一个说明,声称理查德-宾(Richard Bing, 图6)在《约翰-霍普金斯医院公报》上发表了一篇文章,其中心内导管研究表明,"艾森曼格综合征患者的右心室和肺动脉的压力异常高","血液通过心室间隔缺陷从右向左以及从左向右分流。"

图6. 理查德-宾(Richard Bing, 1909-2010),这位心脏病专家首次通过右心导管手术对艾森曼格综合征进行了病理生理学解释。
这些发现使我们能够正确解释艾森曼格综合征发绀的原因。最初,由于从左到右的分流,肺血流量增加,然后肺血管阻力的升高超过了全身,最终分流逆转,从右到左。1951年,Selzer总结说:"艾森曼格综合征的最大特点是存在严重的肺动脉高压"。
这促使伦敦的成人CHD心脏病专家Paul Hamilton Wood(1907-1962)(图7)改变了艾森曼格 "复合体 "的病理生理学定义,即VSD和肺动脉高压与室间分流反转。Wood意识到它可能发生在任何有初始左至右分流的CHD中,并首次提出了Eisenmenger "综合征 "这一名称,意思是指由于肺血管阻力高,并在心房、心室或主动脉肺水平有反向(右至左)或双向分流的肺动脉高血压。随后,"艾森曼格反应 "一词被用来表示肺动脉疾病的发展。

图7. 保罗-汉密尔顿-伍德(Paul Hamilton Wood,1907-1962),英国心脏病专家,他为任何心房、心室或大动脉水平的房间隔缺损引入了艾森曼格综合征这一名称,并并发肺动脉高压和反向或双向分流。此外,他还介绍了肺血管阻力的计算方法(Wood单位)。
反向分流的原因
1958年在梅奥诊所,Jesse E. Edwards(1912-2008;图8)和Donald A.Heath (1928-1997)意识到肺动脉高压是由于毛细血管前的严重阻塞性肺血管疾病和不可逆的变化(图9),并首次提出了六级分类:1级,肺小动脉和小动脉的内侧肥大;2级,内侧细胞增生;3级,内侧洋葱状阻塞性纤维化;4级,动脉瘤;5级,丛状-增生;6级,纤维素坏死。后者可能是动脉破裂和咯血的原因,这也是最初艾森曼格病例的死因。艾森曼格综合征肺动脉高压的组织学基础与特发性肺动脉高压(无分流)和超系统肺血管阻力重叠,是这种肺血管疾病的结果,说明分流逆转。

图8. 杰西-爱德华兹( Jesse E. Edwards,1912-2008)的照片,他首次将艾森曼格综合征的肺血管疾病分类。

图 9. 艾森曼格综合征的阻塞性肺血管疾病。A,显示冬季树脉管系统的肺动脉死后注射。B,内膜阻塞性纤维化。C,动脉瘤和丛状病变。D,纤维蛋白样坏死伴有动脉壁破裂。
Marlene Rabinovitch在70年代和80年代在波士顿儿童医院进行的研究表明,最初肺血流增加的特点是远端毛细血管前动脉的肌肉化,内侧肥大,像持续的胎儿循环。
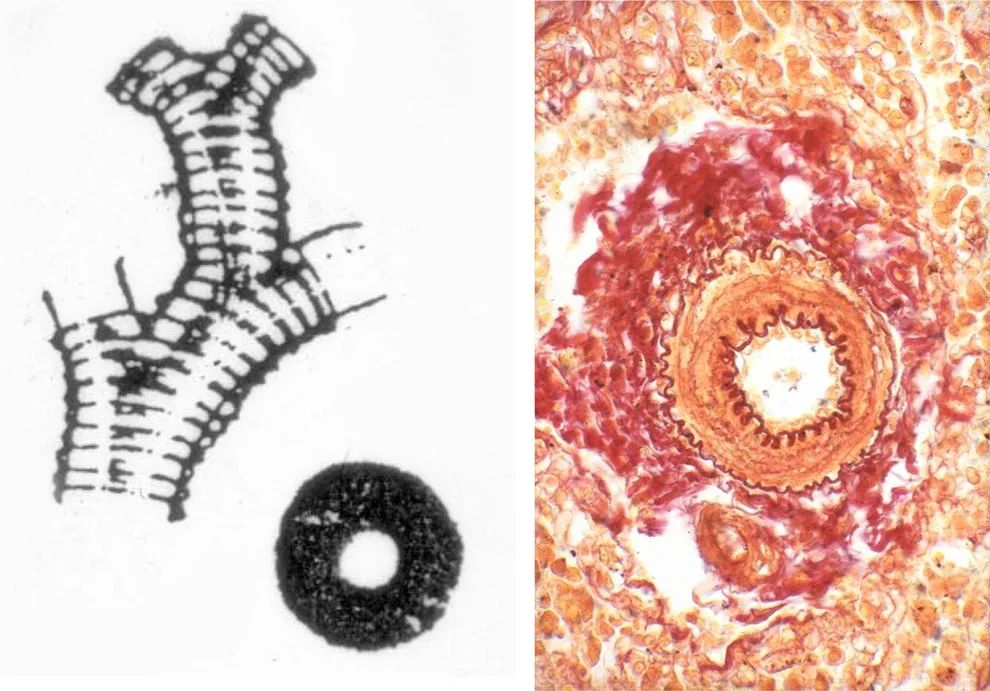
图10. 患有左至右分流的房间隔缺损的肺血管疾病早期阶段的肺动脉血管远端肌肉化。
在70年代和80年代,当外科医生开始在婴儿-儿童时期完成三尖瓣室间隔缺损后的修复工作时,人们发现有些病例在室间隔缺损闭合后断绝心肺旁路,有时右心室会出现急性扩张和低心输出量综合征,就像急性肺源性心脏病,尸检时发现有严重的肺血管病。
在完全性房室间隔缺损中,无论是否有唐氏综合征,所有肺血管阻力>7伍德单位的婴儿都有肺血管病变,而且是不可逆的病变,最早发生在12至24个月大时(图11)。这个信息很明确:尽早预计完全修复,最好是在出生后的头6个月内。

图11. 30名患有先天性心脏病伴房间隔缺损的早期 "不可逆 "肺血管病的婴儿的肺活检。注意手术的高风险,在4级的病例中更严重。
当时在手术前或手术中经常进行肺部活检,以确定修复的指征和预后的目的。平均年龄为两岁的三尖瓣后分流患者的临床病理相关性(图11)显示,在存在不可逆病变(≥3级)的情况下进行修复,会导致死亡率过高。
法洛氏四联症中的艾森曼格综合征的悖论
在法洛氏四联症中,发绀在出生时就存在,是由于肺动脉狭窄,通过VSD的右向左分流所致。相反,在艾森曼格综合征中,发绀是在阻塞性肺血管疾病和反向分流发生后获得的。在法洛氏四联症伴有肺动脉闭锁的情况下,肺动脉供应可能依赖于导管,需要前列腺素治疗和支架来维持导管的通畅,或者是布拉洛克-陶西格分流,以保证动脉血液供应的氧合和生存。然而,也有法洛氏四联症伴有肺动脉闭锁的病例,其中肺动脉循环由来自主动脉的系统-肺动脉旁路供应,向远端肺泡毛细血管供血。这是一个真正的功能性循环,而不是一个营养性的支气管循环。
这些附属的系统-肺动脉被认为是肺部早期胚胎动脉循环的持续存在(节段性动脉),在肺部动脉循环被第六个主动脉弓捕获之前,独立的小循环已经发展。由于主动脉处于高(全身)压力下,出现了主动脉到肺动脉(左到右)的分流,有发生肺血管疾病的危险。患有这种法洛氏四联症的婴儿在出生时是 "粉红色 "的,随着时间的推移会变成 "蓝色"(发绀),因为通过这些分流管发生了左至右的阻塞性肺血管疾病,就像任何房间隔缺损的艾森曼格综合征所发生的那样(图12)。通过组织学的连续切片,我们惊奇地发现,这些准分子在来自主动脉的起源处是肌肉型的,然后在肺希拉处转化为弹性肺动脉,最后转化为肌肉型小动脉和小动脉,就像正常的远端肺动脉血管。如果没有保护肺动脉床的系统串联的病灶狭窄,就可能发生肺血管疾病。

图12. A, 肺动脉闭锁壁室间隔缺损(极端的法洛氏四联症)与肺部侧支循环的方案。B, 法洛氏四联症与肺动脉闭锁的标本,从右心室看:注意四联症(心室间隔缺损、肺动脉闭锁与肺部盲沟、主动脉凌驾于心室间隔之上,以及右心室肥大)。没有动脉导管。C,侧支系统动脉从降主动脉产生,进入肺部庇护所,合并到功能性肺动脉循环。D,远端肺动脉床的阻塞性肺血管疾病。
自然史和治疗
由于艾森曼格综合征的发生,过早死亡是逆转分流患者的严重预后,如果不加以修复。据计算,患者的平均生存概率约为40年,与特发性肺动脉高压相比,这是有利的(图13)。艾森曼格与特发性肺动脉高压相比,预后较好的原因是有房间隔缺损的CHD的 "自然分流"。特发性肺动脉高压会出现右心室衰竭和心脏失代偿的情况,而椭圆窝的穿孔是在特发性肺动脉高压中完成的。

图13. 艾森曼格综合征的自然史比特发性肺动脉高压好得多。
艾森曼格综合征的致命并发症包括肺部(血栓形成、血栓栓塞)、脑部(由于红细胞增多症、悖论和败血症栓塞引起的中风和脓肿)和心脏(心律失常猝死、感染性心内膜炎)。关于心脏死亡,由心室颤动引起的突然心律失常的心脏骤停并不是一种罕见的致命结果机制。
预防的支持性治疗要点
艾森曼格综合征的严重肺动脉高压的药物治疗可通过以下方式积极进行:
1.内皮素受体拮抗剂如波生坦、
2.磷酸二酯酶5*制剂抑**,如西地那非,和
3.两者的结合。
在艾森曼格综合征患者中,在积极治疗肺动脉高压的时代,生存率稳步提高,在七年的随访中,累计死亡率为6%,而在没有这些治疗方法的情况下,累计死亡率为35%(图14)。
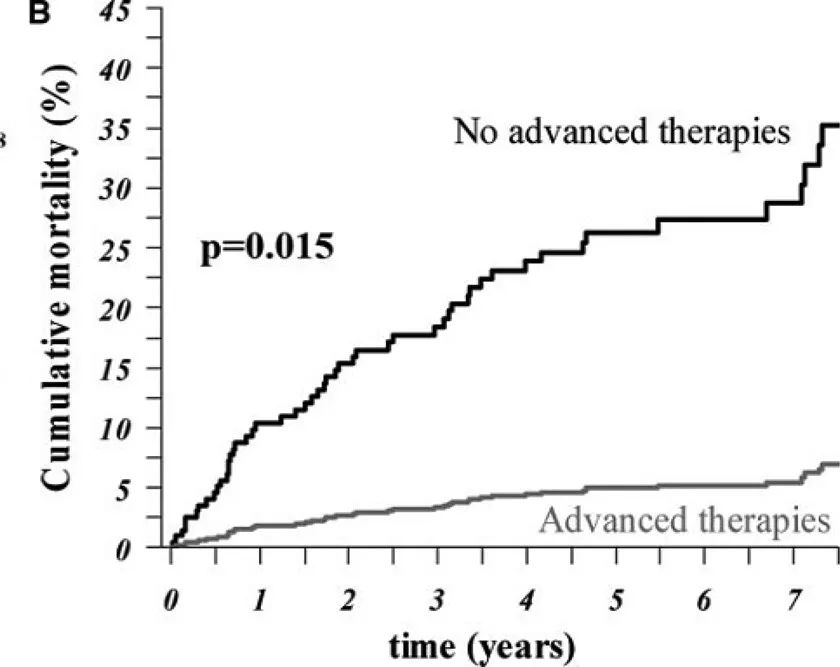
图14. 艾森曼格综合征患者的生存率在晚期治疗中更为有利。
在晚期情况下,移植是一种拯救生命的选择。它可以通过心肺移植或孤立的肺移植(双肺或单肺)来完成,保留原生心脏并修复房间隔缺损。在心肺移植中,被切除的心脏可能被修复,然后捐赠("多米诺 "级联)。肺移植治疗艾森曼格综合征的结果是令人满意的。成年CHD患者心肺或双肺移植后15年的累积存活率为40%,与其他肺实质疾病的肺移植相当(图15)。

图15. 艾森曼格患者GUCH和其他肺部疾病患者的肺移植,无论是心肺还是双肺,其结果都是相似的。
研究结论
维克多-艾森曼格在发表了将他引入医学史的病例报告后,成为弗朗西斯-斐迪南-奥斯堡大公的私人医生,后者于1914年6月28日在萨拉热窝被一名年轻的塞尔维亚恐怖分子杀害。这一事件是第一次世界大战的序幕,也是20世纪上半叶涉及欧洲的一场灾难的开始。如今,当科学为人类健康和福利达到令人难以置信的成就时,乌云笼罩着地球的上空。不幸的是,医学在防止战争和确保全世界和平生活方面仍然无能为力。
参考文献
Thiene G. Eisenmenger Syndrome Revisited. World Journal for Pediatric and Congenital Heart Surgery . 2017;8(6):726-734. doi:10.1177/2150135117727259