本文来自微信公众号:X-MOLNews
饱和碳-碳键在有机分子中普遍存在,并使得有机分子呈现多样的“三维立体”结构。研究也表明,增加手性碳中心的数量有助于提高药物开发成功率。温和条件下高对映选择性饱和碳偶联是有机合成化学研究的焦点和难题,而过渡金属催化的亲电-亲核交叉偶联是高对映选择性构建饱和碳-碳键的有效方法。近年来,不饱和烃*能官**化研究方向也取得了系列进展,为构建饱和碳-碳键发挥了重要作用。
从对映选择性控制模式而言,通过外消旋原料的自由基偶联(图1a)或不饱和双键金属氢化物迁移插入(图1b)都可以获得手性产物。铜、镍等过渡金属在上述两种不对称催化模式中展现出重要的价值。然而,上述反应普遍需要适当的辅助基团实现高对映选择性控制。加州理工学院的Gregory C. Fu教授总结了多种高效的辅助基团( Science , 2017 , 356 , eaaf7230; ACS Cent. Sci. , 2017 , 3 , 692; Angew. Chem. Int. Ed ., 2019 , 58 , 3571),包括Lewis 碱性杂原子基团(如羰基或芳胺的氧、氮原子)或临近的p/π轨道(如芳基、羰基或硼酯基团等)。这些精心设计的辅助基团有效提高了反应对映选择性控制,却也限制了底物的适用范围。
钴催化剂具有廉价、低毒等优点,在均相催化、有机合成中发挥了重要作用。尽管钴催化剂广泛用于碳-碳偶联,但其对映选择性控制仍具挑战(Cobalt Catalysis in Organic Synthesis: Methods and Reactions, Wiley-VCH, 2020 )。发展钴络合物不对称催化体系,有望促进更多不对称饱和碳偶联反应的发现,助力手*药性**物开发。
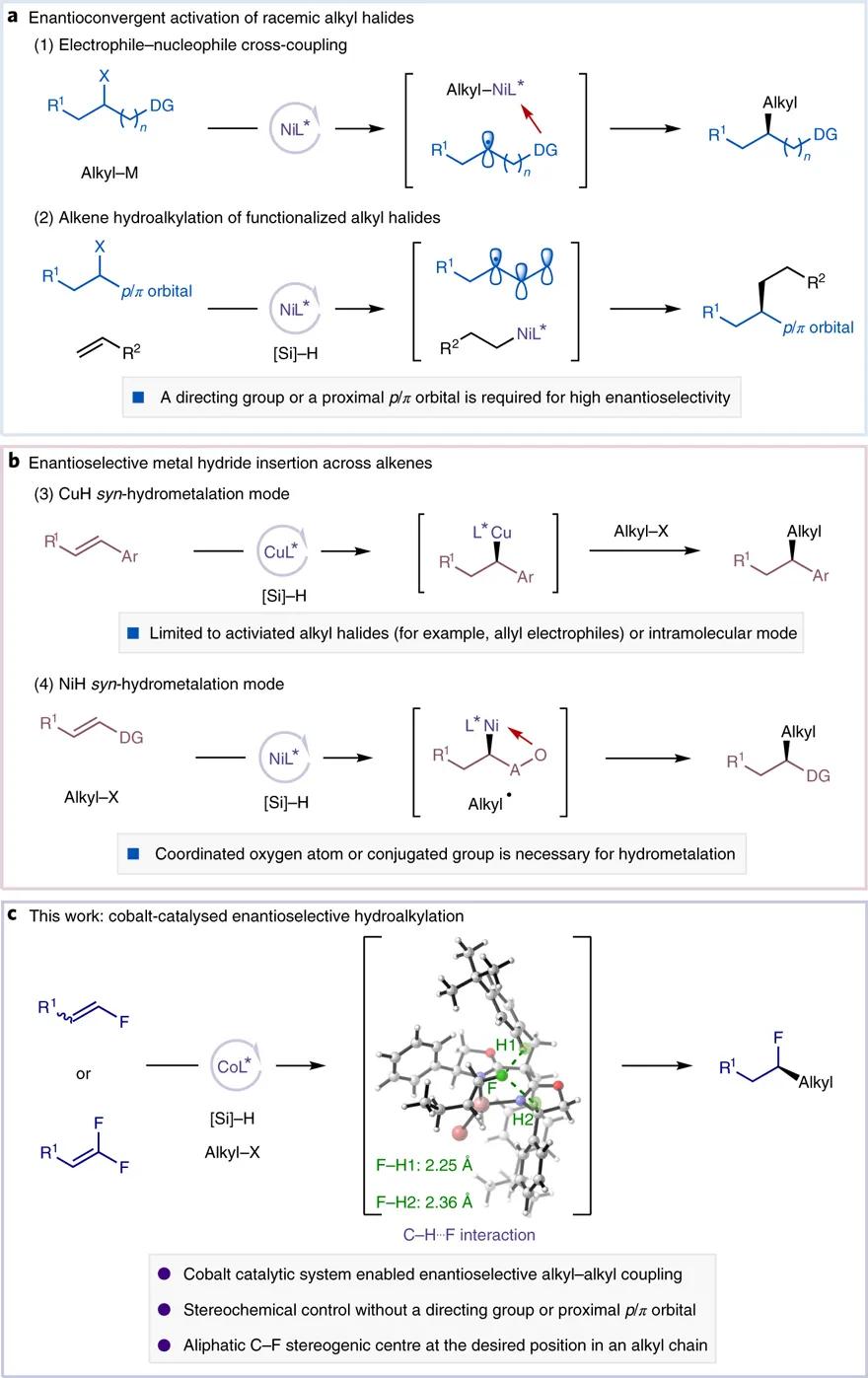
图1. 研究背景及本文工作。图片来源: Nat. Catal.
中国科学技术大学傅尧、陆熹 等长期从事有机化学和绿色化学等领域的研究。基于绿色催化的理念,傅尧、陆熹等提出“烯烃还原偶联”概念,以性质稳定、来源广泛、易于获取的烯烃作为烷基化试剂实现温和条件碳-碳偶联( Nat. Commun ., 2016 , 7 , 11129; J. Am. Chem. Soc ., 2017 , 139 , 12632)。研究团队系统性地开发手性金属镍络合物催化体系,实现系列高对映选择性的烯烃还原偶联反应,为手性胺类/醇类药物分子、含膦/硫/硼精细化学品合成提供了可靠策略( J. Am. Chem. Soc ., 2020 , 142 , 214; Nat. Commun ., 2021 , 12 , 1313; CCS Chem ., 2021 , 3 , 727)。近日,该研究团队开发了 新一代廉价金属钴络合物催化体系,突破了不对称偶联传统辅助基团的限制,以优异的区域和对映选择性实现氟烯烃的氢烷基化 ,为手性氟烷烃合成提供了高效、通用的方案,为不对称饱和碳偶联研究提供了新思路(图1c)。相关成果发表在 Nature Catalysis 上。
条件优化
研究人员以氟烯烃 1a 、烷基碘 2a 为模型底物开展氢烷基化反应的条件优化(图2)。在系统筛选所有反应参数后,研究人员发现在DME(乙二醇二甲醚)溶剂中催化剂CoBr2(DME)和配体( S,S )- L *与硅烷DEMS(甲基二乙氧基硅烷)和碱CsF的组合对反应成功至关重要。在此最优条件下,反应能以85%的分离收率和97%的对映体过量值(e.e.值)生成目标产物 4aa 。图2和补充信息中详细总结了反应参数的影响。此外,研究人员还测试了所使用钴催化剂的原子发射光谱(ICP-AES)并开展了不同金属催化剂的对照实验,排除了钯、铜、镍等其他金属催化剂的残留干扰(详见论文补充信息)。
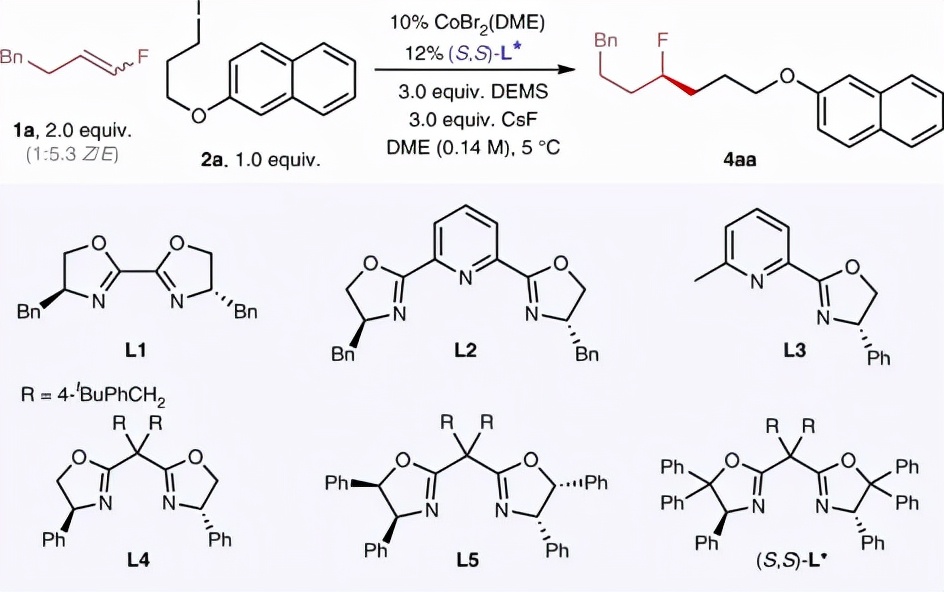
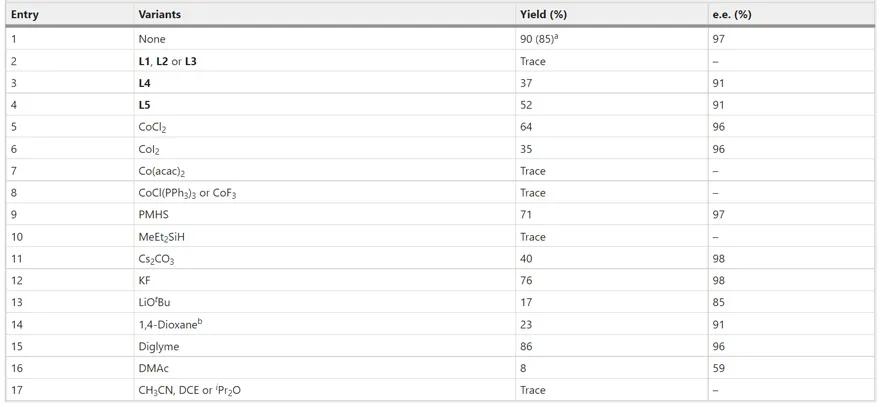
图2. 氟烯烃不对称氢烷基化反应条件优化。图片来源: Nat. Catal.
底物拓展
在最优条件下,研究人员对底物范围进行了考察。如图3所示,该反应对于烷基卤化物适用范围广,产率(40-90%)和对映选择性(88-99% e.e.)高。反应*能官**团兼容性好,如醚( 4aa、4ab )、芳基酯( 4ac )、烷基酯( 4ad )、三氟甲基( 4ae )、氨基甲酸酯( 4ah、4ai )、酮羰基( 4aj )、硫原子( 4af )、氧原子( 4ag、4an )、氮原子( 4ah )、烷基或芳基氯化物( 4ak、4al )和芳基溴化物( 4km )均能兼容。药物设计中,甲基化广泛应用于调控候选药物的生物活性和理化性质。该反应通过碘甲烷及三氘代碘甲烷可以实现甲基及三氘代甲基的引入( 4ao、4lp )。此外,大量例子( 4aq、4ar、4as、4at、4au、4av、4aw、4Cx、4Dy、4az、4aA )表明该反应不需要烷基卤化物*特中**定杂原子导向基团的参与。该反应还可以用于药物分子和天然产物的后期修饰( 4aB、5aB、4aC、5aC、4aD、5aD )。
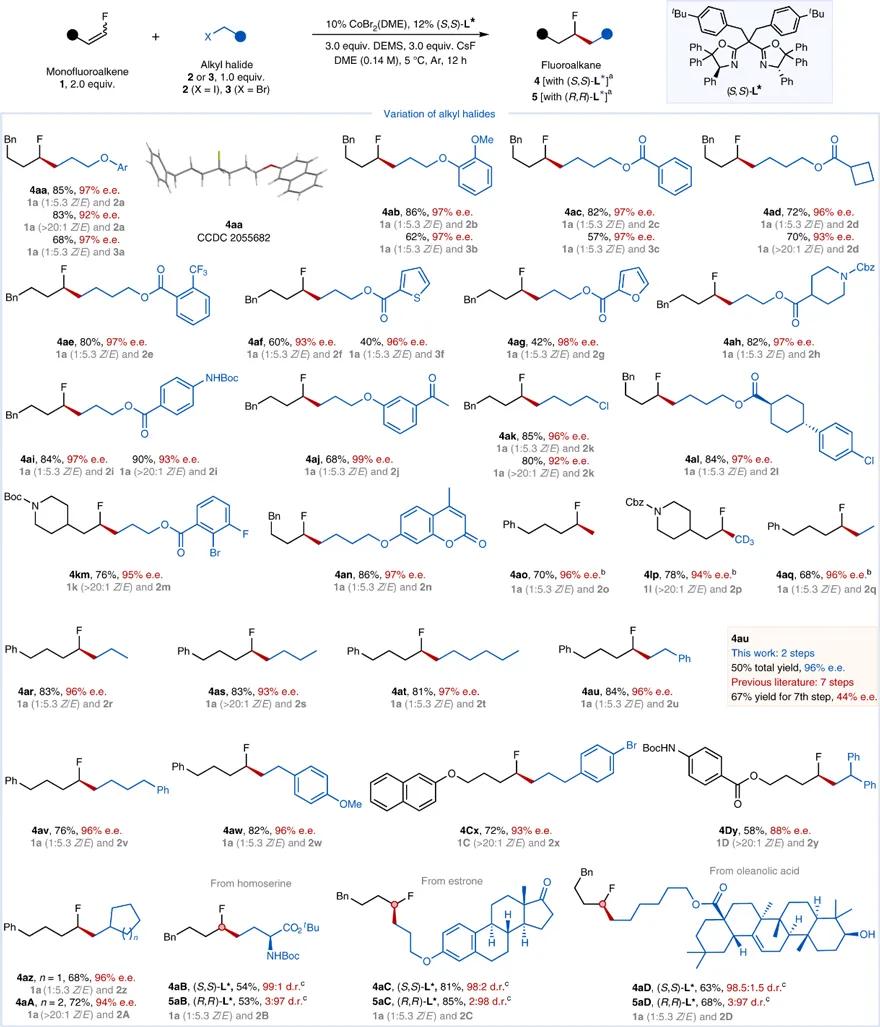
图3. 烷基卤代物的底物拓展。图片来源: Nat. Catal.
研究人员还考察了单氟烯烃的适用范围。如图4所示,各种单氟烯烃同样取得满意的效果。底物 4bi 说明反应的区域选择性和对映选择性受催化剂控制,底物无需配位基团。芳族取代的单氟烯烃也适用于该反应( 4va、4wa、4xF、4yF、4zF )。药物分子吲哚美辛衍生的单氟烯烃 1A 能很容易转化为所需产物 4Aa 。该反应简化Δ11去饱和酶*制剂抑**的合成步骤,进一步展现了反应的合成化学价值。值得注意的是,该反应的产率和对映选择性几乎不受原料单氟烯烃 Z/E 构型的影响。
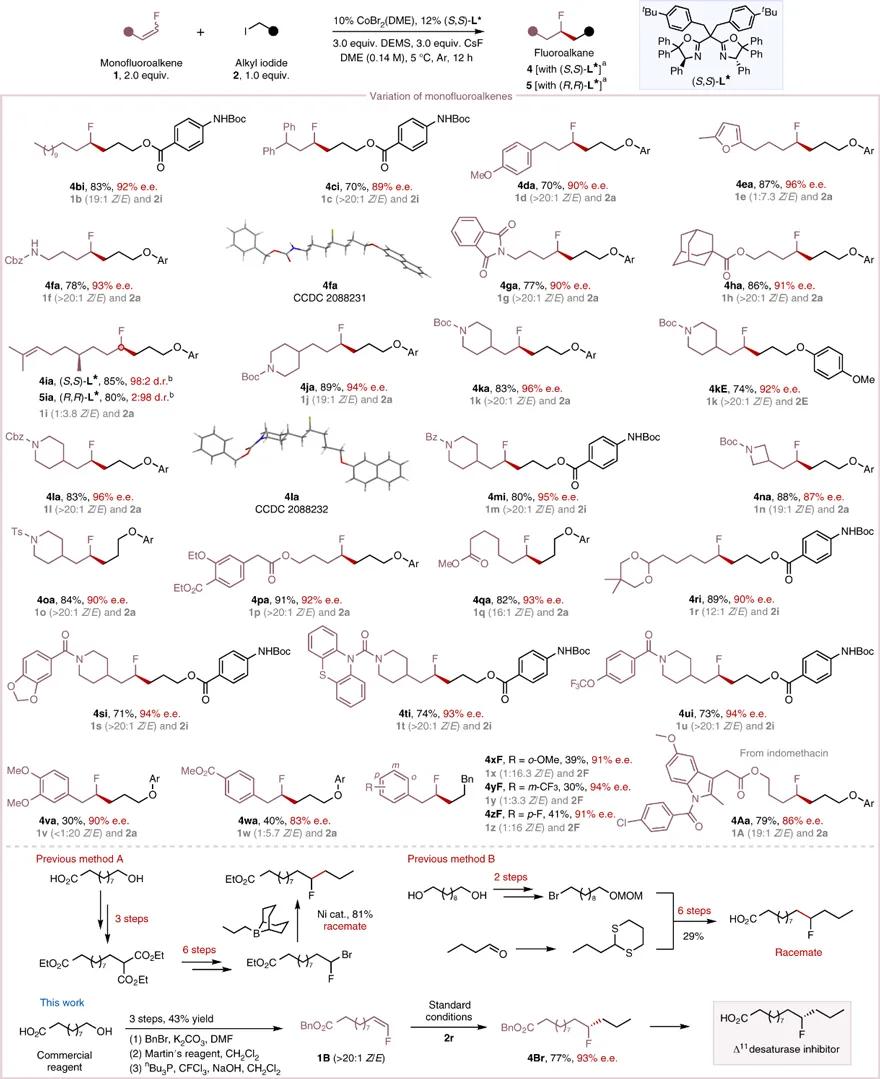
图4. 氟烯烃的底物拓展。图片来源: Nat. Catal.
机理研究
随后,研究人员开展了一系列机理实验。首先,通过自由基钟实验表明卤代烷烃的活化涉及非笼式自由基途径(图5a)。其次,氘代实验结果表明,该反应可能经历Co-H中间体对单氟烯烃的不可逆顺式氢金属化过程,具有高度的区域选择性和对映选择性(图5b)。动力学同位素效应(KIE)实验则表明氢金属化步骤是反应速率限制步骤(图5c)。同时实验还观察到,配体e.e.值和相应产物e.e.值间具有线性关系。研究人员指出,氢金属化步骤可能涉及带有单个双噁唑啉配体的钴络合物。此外,根据紫外-可见光谱(UV-vis)和基质辅助激光解吸/电离飞行时间质谱(MALDI-TOF-MS)的测试,说明CoX2 L *(X = Br或I)是反应体系中主要存在的钴络合物,其可能代表催化剂的静息状态(图5d)。该反应显著的引发期也引起了关注,通过一系类对照实验,研究人员推测引发期生成的Co-H物种是实际催化物种,而不是CoX2 L *(详见论文补充材料)。最后,研究人员还开展了系统的动力学实验。结果表明,烷基碘的活化是快速的非决速步骤,而氢金属化则是反应催化循环的速率限制步骤(详见论文补充材料)。
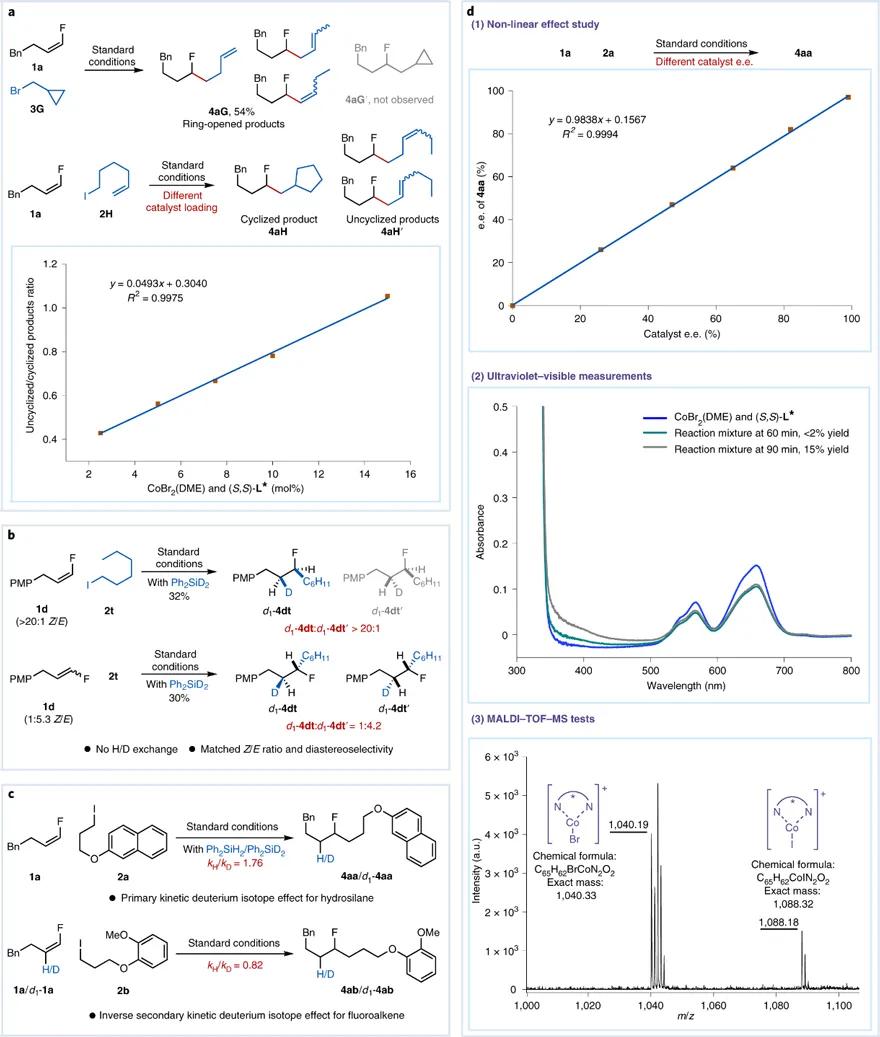
图5. 反应机理探究。图片来源: Nat. Catal.
为了进一步阐明反应的机理过程及选择性成因,研究人员开展了理论计算研究。量化计算结果表明,区域、对映选择性决定步骤和速率限制步骤均为迁移插入(顺式氢金属化)步骤(图6a),与机理实验结果相互印证。其中,钴催化剂自旋交叉过程可以降低该步骤的整体活化能垒。在区域选择性成因的探究上,研究人员认为,相较于空间效应,电子效应起主导作用(图6b)。而对于对映选择性,则归因于不同过渡态下稳定的非共价相互作用(氢-氟相互作用)和不稳定的排斥作用共同决定(图6c)。结合机理实验和量化计算,该反应的可能催化机制如图6d所示。
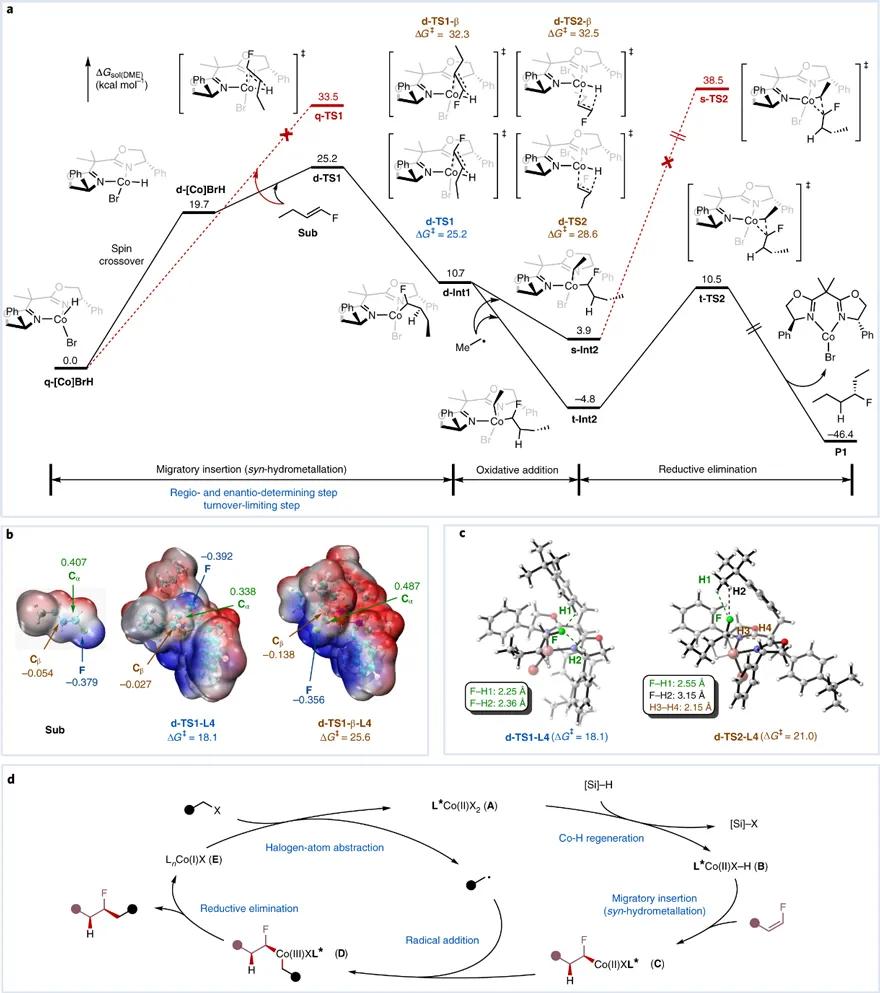
图6. 反应DFT计算探究。图片来源: Nat. Catal.
脱氟氢烷基化
最后,研究人员将该钴催化体系应用于另一大类底物,偕二氟烯烃。如图7a所示,反应条件稍作调整,偕二氟烯烃与烷基卤化物同样可以顺利反应,生成手性单氟烷烃。基于前期“脱氟还原偶联”研究( J. Am. Chem. Soc ., 2017 , 139 , 12632; Chem. Sci ., 2019 , 10 , 809),研究人员提出两条可能的反应途径,P1和P2(图7b)。由于在核磁跟踪实验中观察到单氟烯烃中间体 1a 的生成(图7c),研究人员排除反应途径P1。并推测反应经历途径P2,即偕二氟烯烃首先发生脱氟质子化,随后原位生成的单氟烯烃发生不对称氢烷基化反应得到单氟烷烃。如图7d所示,偕二氟烯烃的脱氟氢烷基化反应同样具有良好的偶联效率(42-68%)和优秀的对映选择性(88-98% e.e.)。
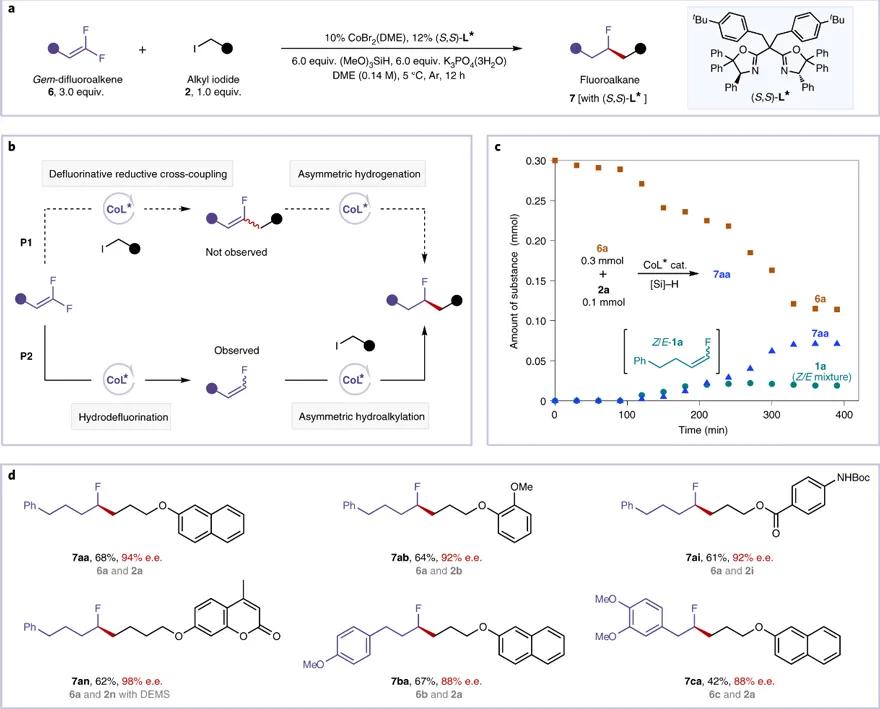
图7. 偕二氟烯烃脱氟不对称氢烷基化反应。图片来源: Nat. Catal.
小结
傅尧、陆熹等开发手性钴络合物催化剂,使用易得的氟烯烃与烷基卤化物为原料,设计配体与底物间的氢-氟相互作用调控反应立体选择性,实现了氟原子邻位手性中心精准构建,突破了不对称偶联中辅助基团结构的局限。初步的机理研究表明,氢金属化是反应的速率限制步骤和立体决定步骤。该研究建立了新型钴-氢催化体系,有望促进更多不对称饱和碳偶联反应的发现,助力手*药性**物开发。
Cobalt-catalysed enantioselective C( sp 3)–C( sp 3) coupling
Yan Li, Wan Nie, Zhe Chang, Jia-Wang Wang, Xi Lu & Yao Fu
Nat. Catal. , 2021 , 4 , 901-911, DOI: 10.1038/s41929-021-00688-w
导师介绍
傅尧
https://www.x-mol.com/university/faculty/14770