
前言
本标准按照GB/T 1.1-2009给出的规则起草。
本标准起草单位:北京医院、上海交通大学医学院附属瑞金医院、中国医学科学院北京协和医院、四川省人民医院。
本标准起草人:彭明婷、谢波、周文宾、李臣宾、谷小林、王学锋、苏薇、赵永强、李焱鑫
1、范围
本标准规定了D-二聚体检测的质量控制要求。
本标准适用于开展D-二聚体检测的临床实验室。
2、规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅注日期的版本适用于本文件。凡是 不注 日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
WS/T 359-2011 血浆凝固实验血液标本的采集及处理指南
医疗机构临床实验室管理办法 卫生部 2006
3、术语和定义
下列术语和定义适用于本文件。
3.1 静脉血栓栓塞症( venous thromboembolism; VTE):
血栓(血凝块)堵塞静脉造成栓塞,包括深静脉血栓形成和肺血栓栓塞症。
3.2 验前概率( pretest probability; PTP):
在明确诊断前,判断患者处于某种临床状态的前期概率。
3.3 阴性预测值(negative predictive value;NPV):
所有阴性结果中真阴性所占的比例。
3.4 敏感度( sensitivit):
实际患病者中检测结果为阳性者所占的比例。
3.5 检测系统(measurement system):
用于检测或评估临床标本中的特定物质存在与否,或对标本中的物质进行定量的一组装置。检测系统包括所有仪器、试剂、配套校准物(适用时)、完成检测所需的其他物品以及操作说明等
4、标本采集和处理要求
4.1 标本采集
4.1.1 依据检测方法的原理和要求,使用血浆或全血标本进行D-二聚体检测。
4.1.2 使用血浆标本时,依照WS/T 359-2011的要求采集标本,推荐使用109 mmol/L枸檬酸钠抗凝的静脉血分离血浆,抗凝剂和血液的比例为1:9。一些D-二聚体检测方法如不受抗凝剂的影响,可使用枸橼酸钠、乙二胺四乙酸(EDTA)或肝素抗凝,实际应用时,应遵循生产厂商说明书的要求。
4.1.3 静脉穿刺采血时,应规范采血流程,以免导致D-二聚体检测结果假性升高。
4.2 标本处理和保存
4.2.1 依照WS/T 359-2011的要求对标本进行处理和保存。特殊情况依照厂商说明书和实验室内部要求。
4.2.2 如使用冰冻血浆标本,检测前将其置于37 ℃水浴快速复溶约5 min(复溶时间与标本体积有关),检测前标本应充分混匀。
4.2.3 血浆标本置于-20~-80℃条件下,保存时间为2年。
4.3 分析前干扰因素
4.3.1 标本有溶血、脂血或胆红素升高,可能导致某些检测方法(如微粒凝集检测法)的结果假性降低,干扰程度取决于检测方法所用的波长和溶血、脂血的严重程度。
4.3.2 高浓度类风湿因子可能对某些检测方法造成干扰,导致检测结果假性升高。
4.3.3 冷球蛋白血症患者血浆中存在异嗜性抗体,可能导致检测结果假性升高。
5 分析方法
5.1 双抗夹心定量检测方法(酶联免疫分析,ELISA):
ELSIA法检测D-二聚体的原理是双抗体免疫测定,其检测结果相对可靠,敏感性高.ELISA法是普遍公认的D-二聚体检测可靠方法,该方法可作为其他常规检测方法比对的标准,主要用于确认其他常规方法检测结果的可靠性。
5.2 微粒凝集定量检测方法:
通过透射/散射比浊原理进行D-二聚体定量检测,是血液凝固分析仪主要采用的检测方法。该方法操作简便,检测快速,可满足常规和急诊标本的检测要求。
5.3 微粒凝集半定量检测方法:
少数实验室使用半定量乳胶凝集法检测D-二聚体。该方法检测快速,适用于高浓度水平D-二聚体检测(如弥漫性血管内凝血、溶栓治疗等),但该方法不适用于VTE的排除性诊断。
5.4 床旁检测(POCT):
POCT检测结果应与可靠的检测方法进行比对,该方法不适用于VTE的排除性诊断。
6检测系统
6.1 实验室宜选择配套的检测系统,用于VTE排除诊断的试剂应有临界值标示并经产品注册审批部门审核批准。
6.2 实验室在选择D-二聚体检测仪器和试剂时,应考虑的因素至少包括:
a) 试剂的预期用途:排除性诊断/辅助性诊断;
b) 检测方法:定性/定量;
c) 可靠的排除诊断临界值以及临界值浓度水平检测结果的精密度;
d) 结果报告时间;
e) 结果报告采用的方式纤维蛋白原当量单位(Fibrinogen equivalent unit,FEU)或D-二聚体单位(D-dimer unit ,DDU),单位选择ng/mL、mg/L或ug/L;
f) 急诊标本及时检测的可行性;
g) 临床安全性。
7性能验证
7.1 性能验证的一般要求:
7.1.1 新的检测系统用于临床检测前,应进行性能验证和校准,包括精密度、线性和正确度等。
7.1.2 仪器使用过程中,每年应对仪器性能进行评审,评审不合格时应进行性能验证。
7.2 批内精密度:
7.2.1 批内精密度验证要求批内精密度以连续检测结果的变异系数(CV)为评价指标,批内精密度应达到试剂生产厂商说明书的要求,同时至少应符合如下要求;正常质控品或血浆标本检测结果CV≤15%,异常质控品或血浆标本检测结果CV≤10%。
7.2.2 验证方法取正常和异常浓度水平的质控品或血浆 样木 ,分别按常规方法至少重复检测10次,计算检测结果的平均值和标准差,按下式计算变异系数。
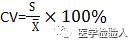
式中:
CV---变异系数
s-标准差;
`X -算术平均值。
7.3 日间精密度:
7.3.1 日间精密度验证要求日间精密度以室内质控结果的变异系数为评价指标,日间精密度应达到试剂生产厂商说明书的要求,同时至少应符合如下要求:CV≤15%。当D—二聚体用于VTE排除诊断时,临界值水平检测结果的日间精密度应 ≤7.5%。
7.3.2 验证方法至少使用2个浓度水平(包含正常和异常水平)的质控品或冰冻血浆,在检测当天至少进行1次室内质控,按批号/按月份计算结果的变异系数。
7.4 线性验证:
7.4.1 线性验证要求线性回归方程的斜率在1±0,05范围内,相关系数r≥0.975 ;标本经稀释后实际检测值与理论值的百分偏差应≤15%,在试剂生产厂商说明书规定的线性范围内应满足上述要求。
7.4.2 验证方法:
7.4.2.1 取1份接近线性范围上限(H)和1份接近线性范围下限(L)的混合血浆,至少制备5份稀释浓度的标本(可按4L、3L +1H 、2L+2H、1L+3H、4H比例配制)。标本按浓度水平由低到高,再由高到低的顺序进行检测并计算2次检测结果的均值。以不同浓度标本的检测结果均值及稀释比例计算各标本的理论值,用实测值与理论值进行回归分析,得出相关系数和斜率。
7.4.2.2 取1份接近线性范围上限的标本,重复检测2次计算结果的均值作为理论值,在仪器上按检测常规标本设定的稀释比例进行稀释和检测,获得样本实际检测值,按下式计算实测值与理论值的百分偏差。

式中:
B:偏差,%
m实测值:实测值
m理论值:理论值
8、校准及校准验证:
8.1 依照厂商说明书规定的程序进行校准,可选择厂商提供的配套校准物或标准物质进行检测结果的校准,也可使用预定标模式进行校准。检测过程中使用的计量器具(如加样器)也应进行校准。
8.2 校准周期及实施条件如下(不限于以下内容):
a) 检测系统用于临床检测前;
b) 更新不同批号试剂后;
c) 室内质控结果显示趋势变化时;
d) 仪器关键部件更换或维修后(必要时);
e) 临床反馈检测结果与症状/体征不相符(必要时);
f) 至少半年1次。
8.3 鉴于D-二聚体检测尚无国际标准品和参考方法,可使用厂商提供的另一批号配套校准物进行校准验证。
9 检测程序依照《医疗机构临床实验室管理办法》、本标准的要求、试剂和仪器生产厂商说明书的要求,制定所在实验室的标准操作程序并予以实施。
10、室内质量控制:
10.1 质控品的选择:
推荐使用配套质控品,也可使用冰冻血浆,使用非配套质控品和冰冻血浆时应与配套质控品同步检测,评价其适用性。
10.2 质控品浓度水平:
至少使用2个浓度水平(正常和异常水平)的质控品。
10.3 质控频度:
据实验室检测标本的数量确定频率,检测当天至少1次。
10.4 质控均值的确定:
质控品检测10 d以上,至少使用20个检测结果计算均值,更换不同批号试剂或仪器进行关键部件维修后,应重新确定质控品的均值;每个批号质控品在使用前,应由实验室通过检测确定均值,制造商规定的“标准值”只能作为参考。
10.5 质控规则:
至少使用 13S和22S规则。
10.6 质控检测结果:
应符合7.3.1的要求。
10.7 质控数据的管理:
质控数据每月总结1次,记录至少保存2年。
10.8 质控记录审核:
实验室负责人或指定负责人应至少每月对室内质控记录进行审核并签字。
11 室间质量评价实验室应参加室间质量评价机构组织的室间质量评价活动,以保证实验室间检测结果的可比性
12 实验室内部结果可比性:
12.1 同一机构内使用多个检测系统时,应尽可能选用同一品牌的检测系统.
12.2 实验室使用同一品牌的多个检测系统时,应定期进行结果比对(至少半年1次)。至少使用20份临床标本(正常和异常标本各10份),每份标本分别使用临床实验室内部规范操作检测系统(使用配套试剂、用配套校准物定期进行校准、检测系统性能良好、规范地开展室内质量控制、参加室间质量评价成绩合格、检测程序规范、人员经过良好培训的检测系统)和被评价检测系统进行检测,以内部规范操作检测系统的结果为标准计算相对偏差,高浓度小平标本的比对偏差应≤10%,低浓度小平标本的比对偏差应≤20%。
13 参考区间及验证要求:
13.1 特定健康人群的D-二聚体结果偏高(如老年人、孕妇和 用产期 妇女),宜对特定人群的参考区间进行验证。
13.2 参考区间验证按照如下步骤实施:
a) 确认所用检测方法的性能符合要求;
b) 至少选择20名健康个体(男女各半),按所在实验室的标准操作程序采集静脉血标本进行检测;
c) 检测结果的离群值检验:首先将检测结果按大小排序并计算极差R,然后分别计算最大值和最小值与其相邻数值之差D;若D/R≥1/3,则将最大值或最小值视为离群值予以剔除,并将余下数据重复上述方法进行离群值检验,直至无离群值为止;
d) 因出现离群值而造成检测结果不足20个时,应重新选择符合要求的健康个体进行补充,以保证20个检测结果不含离群值;
e) 将检测结果与待验证参考区间进行比较。若20个结果超出参考区间的数值不超过10%(2个数据),验证结果符合要求;否则应重新选择20名健康个体再次进行验证,确认验证结果符合要求后,可使用参考区间,否则应查找原因。
14 D-二聚体检测结果用于VTE排除诊断的要求:
14.1 РТP
14.1.1 依据患者临床症状和体征,临床医生应通过标准化的临床评分规则(如Wells和Geneva评分规则)确定患者发生VTE的可能性。
14.1.2 对于低度或中度PTP的患者,D—二聚体检测结果可用于排除VTE,当患者验前概率较高时,不适合使用D—二聚体检测结果进行VTE的排除诊断,需进行影像学检查以明确或排除诊断。
14.2 精密度使用D—二聚体检测进行VTE排除诊断时,临界值水平检测结果的日间不精密度应≤7.5%。
14.3 敏感度和阴性预测值使用D-二聚体检测结果排除VTE,检测结果在临界值浓度水平处的分析性能应符合下表的要求。
表1 用于VTE排除诊断的D-二聚体检测方法的性能要求
敏感度: ≥97%
阴性预测值: ≥98%
阴性预测值95%可信区间的下限: ≥95%
14.4 VTE排除诊断临界值:
14.4.1 建立VTE排除诊断临界值时,被检测D-二聚体的疑似VTE患者应同时使用影像学检查明确或排除诊断,对于排除VTE的患者,应至少在3个月内对其进行跟踪随访,以确认检测结果确实为阴性。
14.4.2 试剂生产厂商应提供多中心研究得出的排除诊断临界值,且该临界值经产品注册审批部门批准适用于相应检测系统,实验室使用临界值前宜进行审核。
15 结果报告:
15.1 D-二聚体定量检测结果有纤维蛋白原等量单位(FEU)和D二聚体单位(DDU)两种报告方式,实验室宜使用生产厂家推荐的报告方式,不宜进行不同报告方式的转换。
15.2 更换不同种类的试剂后,应注意报告方式、参考区间、排除诊断临界值等变化,并与临床进行沟通.
15.3 VTE排除诊断临界值可能与参考区间不相同,当D—二聚体的检测目的为排除VTE时,若VTE排除临界值与参考区间上限值不同,宜报告临界值,且在报告单中注明;此结果用于VTE排除诊断时,仅适用于验前概率低度和中度评分的患者。检测目的不明确时,建议同时报告参考区间及临界值。
参考文献
[1]CLSI:H59-P Quantitative D-dimer for the Exclusion of Venous Thromboembolic DiseaseProposed Guideline, CLSI document. Wayne. PA. Clinical and Laboratory Standards Institute; 2010.
[2] CLSI: H57-A Protocol for the Evaluation. Validation, and Implementation of Coagulometers; Approved Guideline, CLSI document. Wayne, PA: Clinical and Laboratory StandardsInstitute; 2008.