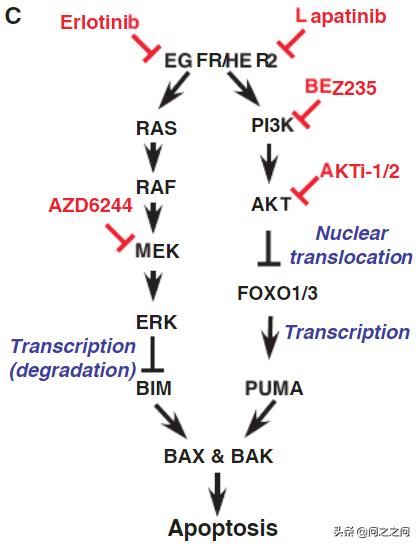
EGFR*制剂抑** 广泛用于临床EGFR突变肺癌患者的治疗,但最终不可避免地出现获得性耐药限制了EGFR*制剂抑**的疗效。在耐药机制中,一代和二代 EGFR TKIs处理后,EGFR T790M “二次突变”占获得性耐药的~50% -60% 。为了克服这一主要耐药机制,已经开发出了三代 EGFR *制剂抑**,接连获批用于临床,包括 osimertinib (FDA,2015)、阿美替尼(2020,中国)和伏美替尼(2021,中国)。osimertinib不仅对T790M获得性耐药有效,而且作为一线治疗优于早期 EGFR TKIs。不幸的是,获得性耐药不可避免地发生而且耐药机制具有异质性,EGFR“二次突变”(C797X,L718X,G724X 等)只在一部分患者中发现(10%-26%)。很明显,仅仅针对 EGFR 突变不太可能治愈 EGFR突变的非小细胞肺癌患者。 诱导肿瘤细胞凋亡是肿瘤靶向治疗成功的关键。 BCL-2家族蛋白是TKI诱导细胞凋亡的关键调节因子。BAX和BAK是线粒体外膜透化(MOMP)的主要效应分子,而BCL-2/BCL-XL/MCL-1对MOMP有抑制作用。BH3s通过直接激活 BAX/BAK 或者使BCL-2/BCL-XL/MCL-1失活来传递上游的凋亡信号来启动细胞凋亡。在凋亡信号中,激活因子 BH3s (BID,BIM,PUMA和NOXA)直接激活 BAX/BAK 诱导BAX/BAK 同源寡聚化导致MOMP的发生。作者团队和其他研究人员先前已经确定BIM 和PUMA 是TKI在体内外*伤杀**EGFR突变NSCLC的关键凋亡效应分子。 具体来说,抑制MEK-ERK信号会诱导 BIM,而抑制PI3K-AKT信号会触发 FOXO1/3核转位,反式激活 PUMA。
Sci Signal. 2013;6(268):ra20.
通常认为,对EGFR TKIs的获得性耐药是通过选择已有的耐药克隆以及“drug-tolerant persisters (DTPs)”(原本致命药物产生暂时耐受能力的癌细胞亚群)的进化而发生的。 随着时间的推移,DTPs可以通过突变或非突变机制获得耐药性。基于这一认识,研究人员假设通过联用增强细胞凋亡的药物可以有效消除癌细胞并减缓耐药的出现。最近,美国纪念斯隆凯特琳癌症研究中心Emily H.Cheng团队通过对化合物库高通量筛选发现, Aurora B激酶*制剂抑**是osimertinib诱导细胞凋亡的强效增强剂,靶向Aurora B激酶能够预防和克服肺癌对EGFR*制剂抑**的耐药 ,相关论文于2021年8月12日以“Targeting AuroraB kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer byenhancing BIM- and PUMA-mediated apoptosis”为题在线发表在《Cancer Cell》杂志上。

Cancer Cell. 2021;39(9):1245-1261.e6.
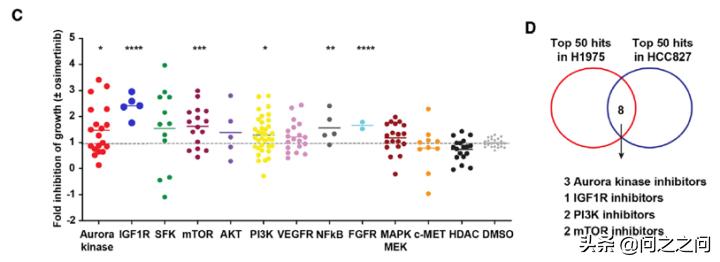
从机制上讲,抑制Aurora B减少BIM蛋白 Ser87磷酸化进而稳定BIM蛋白,并通过 FOXO1/3反式激活 PUMA。 重要的是,上皮间质转变(EMT)引起的osimertinib 耐药会激活 ATR-CHK1-Aurora B信号级联反应,使用相应激酶*制剂抑**可以激活BIM介导的有丝分裂灾难(mitotic catastrophe)导致细胞死亡。也就是说, EGFR*制剂抑**和Aurora B*制剂抑**联用不仅可以有效消除癌细胞,还能克服EMT介导的耐药。 该研究首先确定能增强osimertinib在EGFR突变NSCLC中促凋亡作用的联合用药策略,通过对一个包含~1000个化合物的定制化合物库进行高通量筛选,在H1975和HCC827细胞上检测发现3种Aurora激酶*制剂抑**、1种IGF1R *制剂抑**、2种PI3K *制剂抑**以及2种mTOR *制剂抑**可以协同抑制细胞增殖。进一步在PC9和PDC细胞ECLC26(L858R)上验证研究证实 Aurora激酶*制剂抑**是osi诱导不同EGFR突变的NSCLC细胞株凋亡的强效促进剂。
Cancer Cell. 2021;39(9):1245-1261.e6.
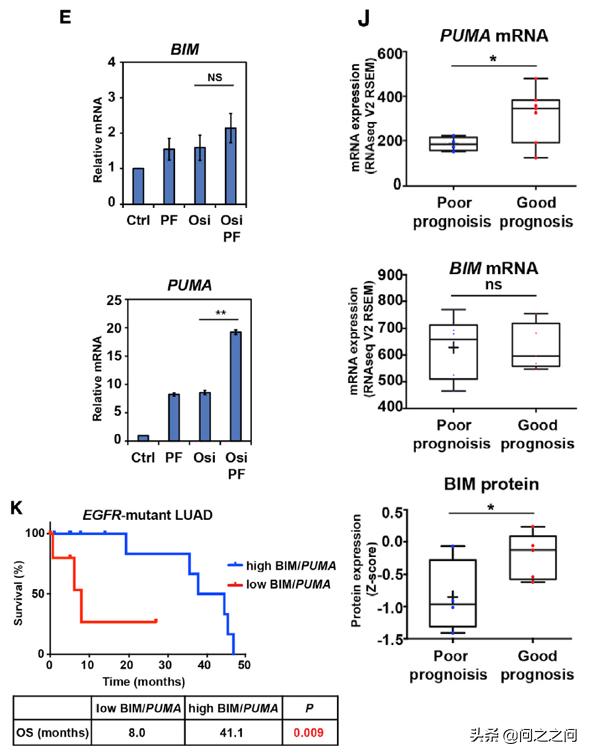
Western检测发现,Aurora激酶*制剂抑**PF03814735(PF)与osi联用后,BIM和PUMA蛋白在蛋白水平显著上调,伴随着AKT和FOXO1/3磷酸化抑制。qRT-PCR检测发现,PF介导的PUMA的表达增加主要发生在转录水平,而BIM mRNA没有明显变化。这些数据表明, PF和osi协同抑制AKT,从而减少FOXO1/3磷酸化,导致FOXO1/3介导的 PUMA 诱导表达。 而PF03814735是Aurora激酶*制剂抑**,对Aurora A 和Aurora B激酶没有选择性。研究人员利用siRNA选择性敲除 AURKA 或 AURKB 基因后发现,敲低 AURKB 基因大大增强了osi诱导的细胞凋亡,并且重现了PF诱导的BIM和PUMA上调,而敲低 AURKA 基因影响甚微。为了进一步探讨 BIM/PUMA 与临床结果之间的相关性,研究人员评估了TCGA数据中EGFR突变的LUAD患者数据发现,肿瘤组织中PUMA mRNA 的表达显著增高的患者预后较好,而其BIM mRNA 的表达没有显著差异。值得注意的是,预后良好的肿瘤患者的 BIM蛋白水平显著高于预后不良患者,提示EGFR信号对BIM蛋白稳定性存在调控作用。与这些发现一致的是,BIM蛋白或PUMA mRNA 表达低的患者,其总体生存期明显缩短(8.0 vs. 41.1个月)。
Cancer Cell. 2021;39(9):1245-1261.e6.
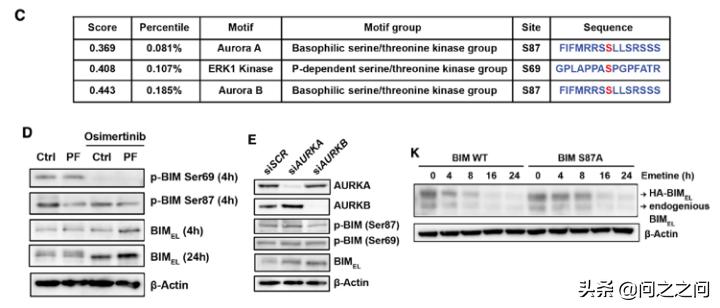
由于PF和siRNA介导的BIM诱导主要在蛋白质水平,研究人员进一步研究发现PF 和osi联用影响了BIM蛋白稳定性。已有文献报道ERK会影响BIM磷酸化进而影响BIM蛋白稳定性,但PF单用对ERK磷酸化影响较小。于是,研究人员猜测Aurora B可能直接磷酸化BIM引起其磷酸化依赖的泛素化降解。利用SCANCITE 4.0软件分析和实验验证发现,生物学和化学手段抑制AURKB会降低BIM S87磷酸化,导致BIM稳定。
Cancer Cell. 2021;39(9):1245-1261.e6.
到此,研究人员证明了EGFR和AURKB联合抑制可以有效消除TKI-naïve肿瘤细胞,紧接着提出疑问,这种联合策略对osi耐药细胞是否同样有效。 通过构建osi耐药细胞株H1975R和ECLC26R,RNA-seqGESA分析发现EMT基因上调是主要原因(蛋白水平E-cadherin表达减少、vimentin表达增加)。有趣的是,H1975R和ECLC26R对Aurora激酶*制剂抑**PF和MLN8054高度敏感。从机制上分析,PF单用可以诱导BIM/PUMA上调,且轻微抑制AKT磷酸化;而osi不能诱导H1975R和ECLC26R细胞凋亡,既没有减少ERK/AKT磷酸化,也没有诱导BIM/PUMA。 为了明确EMT和osi耐药以及Aurora激酶*制剂抑**敏感性之间的因果关系 ,研究人员试图通过干扰EMT关键调节因子来诱导H1975发生EMT。 同时敲除FOXA1/2和过表达ZEB1 (H1975FZ)重现了EMT特征 (E-cadherin减少、vimentin增加) ,H1975FZ对osi耐药且对Aurora激酶*制剂抑**敏感,这些数据表明osi耐药细胞的EMT特征与其对Aurora激酶*制剂抑**的敏感性是有关的。进一步研究发现,多种Aurora激酶*制剂抑**以及siRNA敲低AURKB可以诱导osi耐药的EMT细胞凋亡,而AURKA选择性*制剂抑**MK5108以及敲低AURKA不能诱导凋亡。为了明确osi耐药的EMT细胞对AURKB*制剂抑**高度敏感的分子机制,研究人员分析RNA-seq数据发现,在H1975R和ECLC26R中ATM和ATR上调,并通过免疫印迹验证。ATR通过其下游效应器CHK1调节AURKB进而控制细胞分裂是已知的。相应地,在H1975R和ECLC26R细胞中发现CHK1S345磷酸化增加,而且AURKB自磷酸化显著增加,说明在osi耐药EMT细胞中AURKB是激活的。 这些结果表明ATR-CHK1-AURKB信号在osi耐药EMT细胞中被激活 ,且在H1975FZ细胞中重现了这一结果。
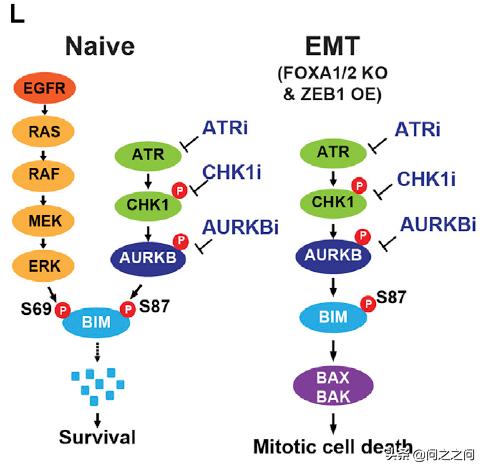
Cancer Cell. 2021;39(9):1245-1261.e6.
最后,研究人员在小鼠异种移植模型中证实osi和PF联合比osi单用体内疗效更好,无论是亲本H1975,还是通过EMT对osi获得性耐药的H1975R模型。在两种PDX模型上,osi和PF联合同样有效地抑制肿瘤生长。总之, 这些数据表明,无论是否有EMT,osi和PF联用对于treatment-naïve或者osi耐药的EGFR突变NSCLC都是一种有效的治疗策略。
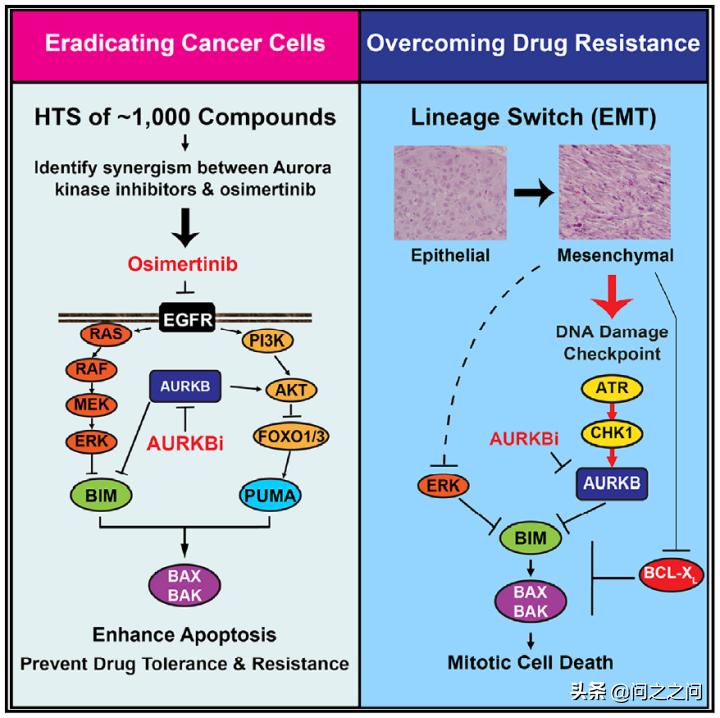
Cancer Cell. 2021;39(9):1245-1261.e6.
值得注意的是,AURKA也被报道介导EGFR三代*制剂抑**耐药 (Nat Med 25, 111–118 (2019)) ,而在本文研究中AURKB激活相较AURKA更明显。同时也有其他研究表明AURKB激活是一代和三代TKIs获得性耐药机制,并伴有磷酸组蛋白 H3(pH3)水平增加 (Nat Commun 10, 1812 (2019)) 。
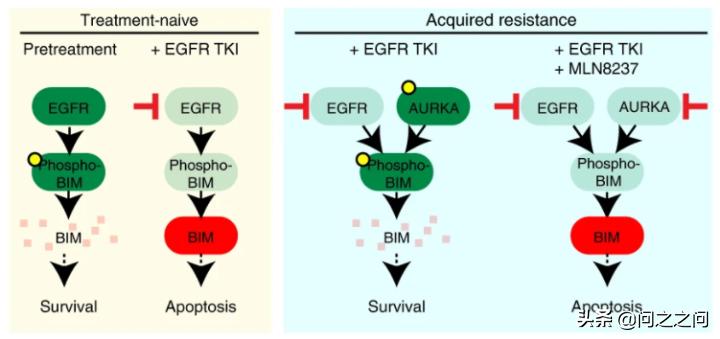
Nat Med. 2019;25(1):111-118.
参考文献:
Tanaka K, Yu HA, Yang S, et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 2021;39(9):1245-1261.e6.
Shah KN, Bhatt R, Rotow J, et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med. 2019;25(1):111-118.
Bertran-Alamillo J, Cattan V, Schoumacher M, et al. AURKB as a target in non-small cell lung cancer with acquired resistance to anti-EGFR therapy. Nat Commun. 2019;10(1):1812.