撰文 | Qi
Angelman综合征 是一种由导致泛素蛋白连接酶E3A ( UBE3A ) 基因的母体等位基因功能丧失或沉默的突变引起的神经遗传性疾病,患者会出现包括发育迟缓、严重的运动和认知缺陷以及癫痫,目前尚无有效治疗策略。在神经元中,UBE3A基因受遗传印记的影响,即母系等位基因表达而父系等位基因不表达 【1, 2】 ,因此,目前正在探索的策略之一就是重新激活父系等位基因。已知父本UBE3A等位基因被UBE3A反义 (UBE3A-AS) 转录本的表达抑制,几项研究表明利用Gapmer 反义寡核苷酸 ( ASO ) 抑制小鼠和人类Ube3a-AS/UBE3A-AS转录本的表达可重新激活神经元中父本Ube3a/UBE3A等位基因的表达,恢复Ube3a在大脑中的表达可改善Angelman综合征小鼠模型的神经功能缺损 【3, 4】 ,但目前尚不清楚这些ASOs是否能直接转化为临床开发。
为此,来自美国德州农工大学的 Scott V. Dindot 团队在 Science Translational Medicine 杂志上发表了一篇题为 An ASO therapy for Angelman syndrome that targets an evolutionarily conserved region at the start of the UBE3A-AS transcript 的文章,他们 描述了针对Angelman综合征的、靶向UBE3A-AS转录本进化保守区域的ASO疗法的开发,证明该ASO可以精确有效抑制UBE3A-AS转录,并重新激活父系UBE3A等位基因的表达 。这些发现支持将这种针对Angelman综合征的治疗策略推广到临床开发 (ClinicalTrials.gov,NCT04259281) 。

UBE3A-AS转录本来自于父系表达的转录单位SNHG14 (剪接和 3'-切割/聚腺苷酸化) ,大部分关于UBE3A印记的知识都是基于对小鼠的研究,但小鼠和人类SNHG14转录单元存在显著差异 (比如小鼠~1000 kb而人类~700 kb) ,人鼠UBE3A-AS/Ube3a-AS转录本的保守程度从未得到充分探索。通过对人鼠转录本的遗传结构和序列分析发现两者之间没有序列或结构同源性,因而无法确定适合检测两个物种的ASO序列。随后,研究人员开发了一种靶向的比较基因组学方法来识别宿主基因远端的ASO靶区,该区域可以精确有效地在UBE3A的3'端之前终止UBE3A-AS的转录,结合系统发育学的研究,作者发现E-3和SNORD109B外显子的序列和剪接在整个胎盘哺乳动物进化过程中都非常保守,但在啮齿类动物中丢失了。
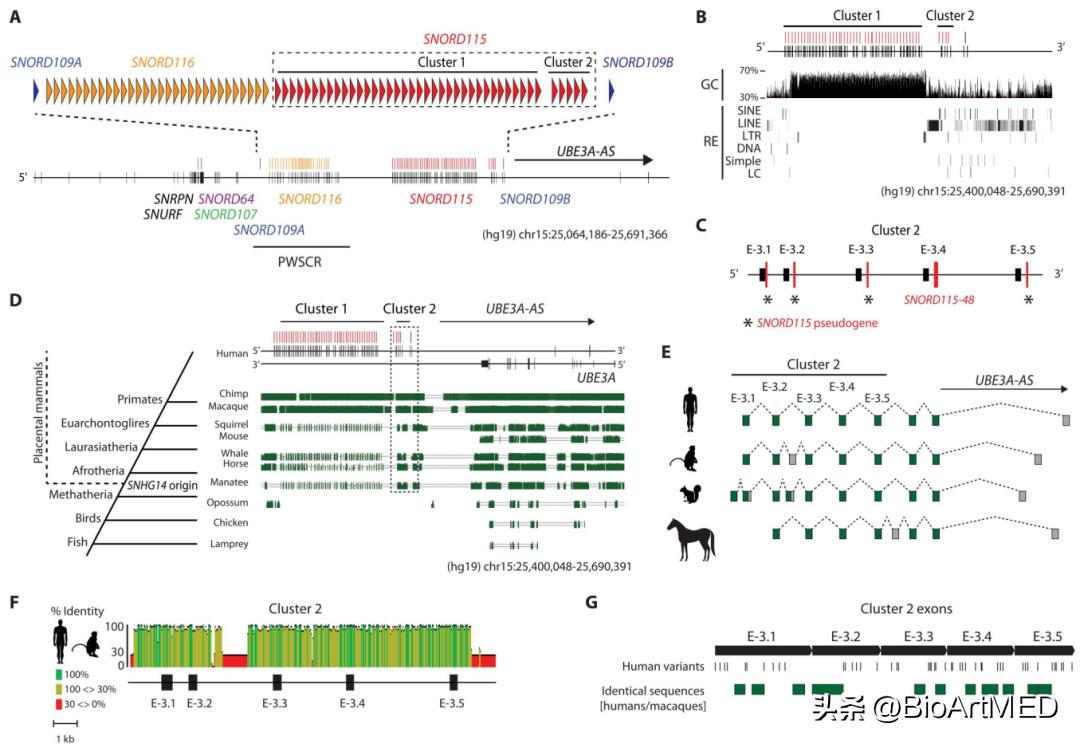
图1. UBE3A-AS转录本是进化保守的。
研究人员将SNORD115划分为两个簇 (如上图所示) ,假设靶向簇2的Gapmer ASO 会在父本 UBE3A 等位基因的 3'-末端之前特异性有效地抑制 UBE3A-AS 的转录。为了验证这一点,作者设计了6个靶向簇2中E-3外显子的ASO,其中包括3个靶向人和食蟹猴相同序列的ASO,随后用iPSC神经元检测ASO的体外作用。ASO处理后,作者用RT-PCR对UBE3A-AS的表达进行量化,每种ASO都能以剂量依赖性的方式显著抑制UBE3A-AS表达,但对位于上游的其他RNA几乎没有影响,说明这些ASO的特异性,在此基础上作者对这些ASO进一步优化以增强效力。为了确认经ASO处理的iPSC神经元中父系UBE3A等位基因的再激活情况,作者利用等位基因特异性数字液滴RT-PCR (ddRT-PCR) 测定来量化RNA丰度。结果显示,其中一种ASO (4.4.PS.L) 以剂量依赖性方式抑制UBE3A-AS表达并使父本UBE3A表达增加622%。为了进一步证实这一点,作者处理了衍生自Angelman综合征患者的iPSC神经元,观察到每种ASO都会抑制UBE3A-AS转录物的表达,并增加Angelman综合征神经元中父系UBE3A等位基因 (RNA和蛋白质) 的表达。
前面的分析表明食蟹猴SNHG14转录本的序列和加工在人类中高度保守,为了评估ASO的体内药理学特性,作者通过腰椎鞘内注射将四种不同的ASO (代表不同的靶序列、骨架设计和RNA修饰) 以不同剂量分别施加给食蟹猴,利用RT-PCR检查UBE3A-AS转录物在11个CNS区域中的表达情况。在对ASO保持良好的耐受性的同时,作者发现ASO抑制不同CNS区域的UBE3A-AS表达,其中腰脊髓、大脑皮层 (运动、额叶和颞叶) 和海马体的影响最大。两种ASO (4.4.PS.L和4.4.PO-1.L) 以剂量依赖性方式抑制UBE3A-AS表达,在几个CNS区域实现超过85%的减少,这些发现说明ASO、CNS区域和给药方案都会影响治疗效果。为了确认父本UBE3A等位基因的重新激活,作者使用等位基因特异性ddRT-PCR分析发现父本UBE3A在几只动物的腰椎脊髓、大脑皮层和海马体中的表达增加,在六只动物的脊髓中实现了完全双等位基因表达。
总的来说, 这项工作描述了用于治疗Angelman综合征的ASO的发现、开发和表征,结果的有效性支持将该分子疗法推进到临床开发,其中4.4.PS.L的1/2期临床试验目前正在进行中 (NCT04259281) 。
原文链接:
http://doi.org/10.1126/scitranslmed.abf4077
制版人:十一
参考文献
1. T. Kishino, M. Lalande, J. Wagstaff, UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73 (1997).
2. U. Albrecht, J. S. Sutcliffe, B. M. Cattanach, C. V. Beechey, D. Armstrong, G. Eichele, A. L. Beaudet, Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet . 17, 75–78 (1997).
3. J. M. Wolter, H. Mao, G. Fragola, J. M. Simon, J. L. Krantz, H. O. Bazick, B. Oztemiz, J. L. Stein, M. J. Zylka, Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 587, 281–284 (2020).
4. C. Milazzo, E. J. Mientjes, I. Wallaard, S. V. Rasmussen, K. D. Erichsen, T. Kakunuri, A. S. E. van der Sman, T. Kremer, M. T. Miller, M. C. Hoener, Y. Elgersma, Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight 6, e145991 (2021)