
阅读此文前,诚邀您点击一下 “关注” , 方便您随时查阅一系列优质文章,同时便于进行讨论和分享,感谢您的支持~

文|大菠萝
编辑|WEIXIAO
一、引言
分子氢代表了未来能量转换方法的理想清洁燃料,因此,可持续氢循环的策略引起了人们的极大兴趣。
[FeFe] 氢化酶是一种有价值的酶,可通过称为 H 簇的催化中心催化双氢的可逆演化(图 1,上图)。 这个复杂的金属位点由一个类似铁氧还蛋白的 [4Fe4S] 簇组成,它通过半胱氨酰硫醇盐共价连接到一个独特的 [FeFe] 中心。 [FeFe] 部分的两个铁原子由仲胺二硫醇配体氮杂二硫醇 (adt) 桥接,并由总共两个 CN -和三个 CO 配体配位。

5个远端铁原子 Fe d(相对于 [4Fe4S] 簇)有一个空位配位点,它应该参与底物结合和转化。
在稳态条件下,H-团簇的至少三种潜在催化中间体可以通过红外 (IR) 光谱清楚地区分,这能够探测双原子配体的结构敏感的 CO 和 CN 伸缩振动(图 1,底部) , 氧化的 H 团簇 (H ox ) 的特征在于[FeFe] 中心的[Fe I Fe II ] 混合价基态和氧化的 [4Fe4S] 2+团簇。

共振拉曼 (RR) 光谱是一种强大的技术,可以详细了解选定的金属-配体振动和金属蛋白的基本分子坐标,例如 氢化酶。
在本研究中,我们首次使用该技术在不同氧化还原条件下探测 [FeFe] 氢化酶的 H 簇。 在这里,我们选择来自绿藻莱茵衣藻的 HydA1作为光谱研究的理想模型系统,因为它只包含 H 簇,没有可能掩盖光谱的额外辅因子。

此外,holo-HydA1 可以在体外成熟通过添加合成的 [FeFe] 复合物从“载脂蛋白”(仅包含 [4Fe4S] 簇)中分离出来。
这允许对本地和非本地 H 簇衍生物进行表征,并且可以单独探测相应的辅因子构建块。在目前的工作中,我们通过 RR 光谱表征了这些前体形式和体外成熟的全息 HydA1(参考文献 18)。 基于这些研究,我们提供了对 H 簇子位点及其相互作用的见解,并将结果与 [FeFe] 氢化酶的催化机制联系起来。

二、实验细节
IscR 缺陷型大肠杆菌菌株 BL21 (DE3) ΔiscR 用于 [FeFe] 氢化酶 apo-HydA1 的异源表达,如前所述,仅含有 [4Fe4S] 立方烷簇。通过HIS 6 -Tag IMAC厌氧纯化氢化酶,并将主要洗脱级分浓缩至 2 mM。

Holo-HydA1(adt) 及其无催化活性的类似物 holo-HydA1(pdt) 是通过体外掺入相应的 [FeFe]-adt 和 [FeFe]-pdt(丙烷-二硫醇桥联)复合物制备的,浓度为 10 倍摩尔根据 Esselborn 及其同事在 2013 年描述的程序,将过量的酶转化为 apo-HydA1。
从这种制备开始,通过用分子氢(超还原态)和一氧化碳(CO 抑制态)冲洗样品 30 分钟或通过添加 2 倍摩尔过量(氧化态)的硫堇。

分离的全息-HydA1(pdt) 和载脂蛋白-HydA1 的氧化是根据针对全息-HydA1(adt) 所描述的程序,通过与硫堇孵育来完成的。 为了正确比较从不同 HydA1 样本记录的 RR 数据,手稿图 3中描述的光谱按如下所述进行了归一化。
使用硫堇氧化的全息-HydA1(adt) 的 RR 光谱作为参考('ox',图 3A),差分光谱Δ = f × s - ox(图 3B-D中的黑色痕迹)计算了所有基线校正之前的其他光谱(对应于图 3B-D中的彩色迹线)。

在每种情况下,缩放因子f的调整方式使得相应的差异光谱反映s之间的定性差异和 ox 而不仅仅是 Fe–CO/CN 中心正常模式 (400–700 cm -1) 的光谱区域中整体强度的变化。
我们还评估了蛋白质的(非共振激发的)苯丙氨酸侧链模式的带强度(在约1005 cm -1,见 ESI 1 †)。 缩放光谱3A–D的比较显示相对于该内部标准只有很小的强度变化(小于 10%),从而证实上述归一化程序允许正确评估相对条带强度。

将含有 50% v/v 甘油的 HydA1 样品 (1.1 mM) 转移到厌氧手套箱内带有 CaF 2窗口(光程长度 = 55 μm)的 气密定制夹层池中。
使用配备有液氮冷却的 MCT 检测器的 Bruker IFS28 FTIR 光谱仪记录红外光谱。在室温下测量光谱后,使用定制的液氮低温恒温器将样品室冷却至 80 K。 在使用 LED 面板 ( λ max = 460 nm)持续照明之前、期间和之后记录了低温光谱。随后升温,再次记录室温光谱,以检验所有样品测量后的完整性(见ESI 2†)

三、结果与讨论
为了揭示光谱标记区域,我们首先比较合成的 [FeFe]-adt 复合物、含有 [4Fe4S] 簇的载脂蛋白和体外成熟的 holo - HydA1(adt) 的 RR 特征(图 2 ),在 300 和 400 cm -1之间的区域中的带预计主要来自 Fe-S 模式。
对于全息-HydA1(adt),与原型立方烷簇和“载脂蛋白”的 RR 光谱的比较表明,这些条带主要归因于 [4Fe4S] 部分,而来自 [FeFe ] 子站点可能很小。
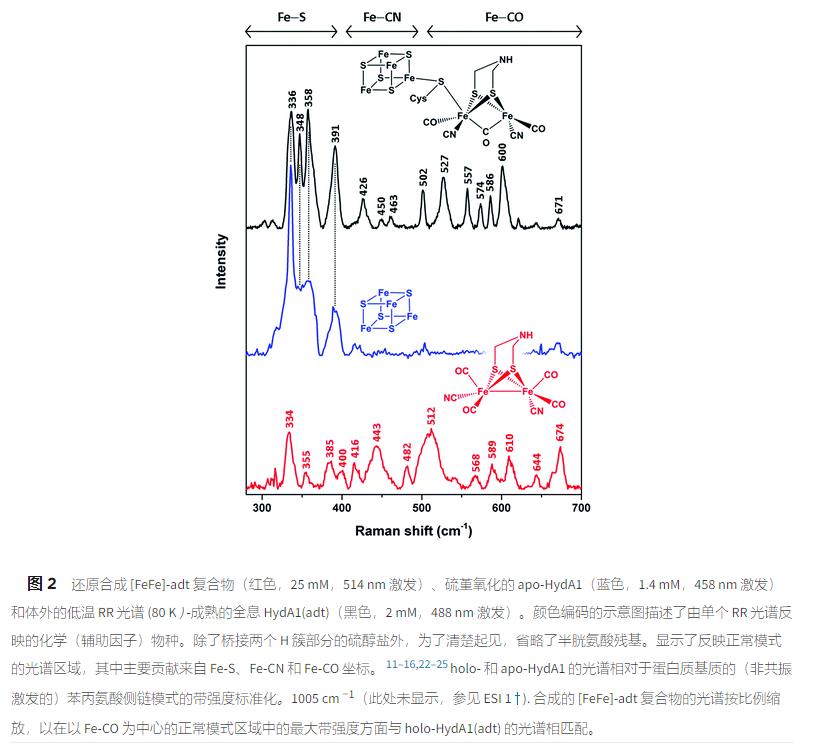
这种效果对于 [FeFe]-adt 复合物尤为明显,它在掺入“apo 蛋白”后显示正常模式频率、带宽和相对强度的明显变化。
这些变化主要归因于与蛋白质基质的相互作用,这对其在催化质子还原中发挥作用所需的辅因子结构施加了限制。 同样的解释可能适用于 holo-HydA1(adt) 中 [4Fe4S] 簇的两个尖锐带,位于 348 和 358 cm -1处,这在“载脂蛋白”的光谱中未被解析。

接下来,我们记录了体外成熟的全息-HydA1(adt) 制剂的 RR 光谱,这些制剂富含不同的 H 簇氧化还原状态(图 3,左和中)。光谱是用 488 nm 激发获得的,它为两个金属位点提供了共振增强。通过在相同温度 (80 K) 下进行的互补红外测量(图 3 ,右)来评估单个氧化还原状态的贡献。
IR 光谱是在黑暗中(浅色线)和蓝光(460 nm,饱和色线)照明期间记录的,后者模仿 RR 测量的条件。
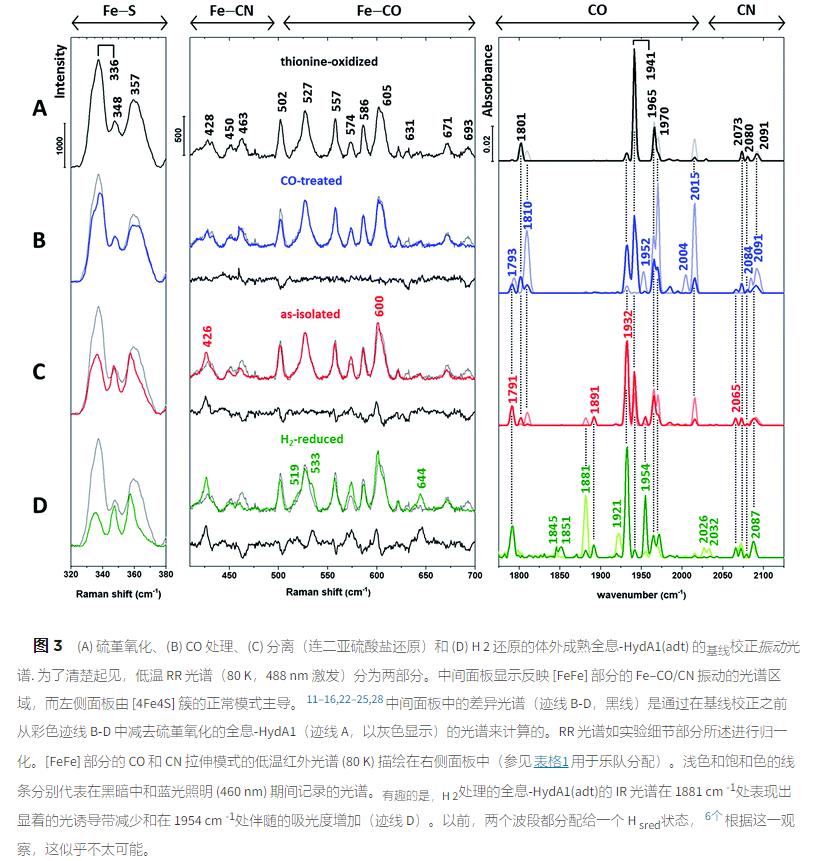
HydA1 对氧气高度敏感,因此在还原条件下厌氧分离并在连二亚硫酸盐存在下储存(在下文中称为分离状态,图 3C)。分离酶与硫堇(图 3A)和氢气(图 3D)的孵育分别能够富集氧化酶和(超)还原酶。
此外,从经 CO 处理的分离酶(图 3B)获得光谱数据,以确定 CO 抑制状态的可能贡献。

因此,我们得出结论,H red的转化涉及一个额外的基本步骤,它能够在 RR 测量期间实现 H' red的动力学稳定。
这一步被认为是质子转移到附近的碱基(最有可能是半胱氨酸 C169),与 [NiFe] 氢化酶中催化中间体的光反应一致。 [FeFe] 部分的桥接 adt 配体是最有可能的质子供体,因为它应该在 H red而不是 H ox中被质子化。
考虑到 H 的相似 [FeFe] RR 签名ox和 H' red(图 3,中间),因此我们得出结论,adt 配体在后一种物种中也被去质子化。

因此,IR 光谱(图 3,右)的显着变化反映了不同处理样品中的各种氧化还原态分布,与 [FeFe] 部分的相应 RR 光谱的相当小的变化形成对比(图3,中间)。为了解决这种差异,我们将注意力转向 [4Fe4S] 簇的 RR 光谱区域特征(图 3,左)。
该区域中的带在很大程度上可以被视为氧化 [4Fe4S] 2+状态的标记,因为对于还原的 [4Fe4S] 1+状态,预计很少或没有共振增强(参见 ESI 1 †)。特别是,氧化样品中最突出的条带(图 3A,左)在 336 cm -1处是氧化的 [4Fe4S] 2+簇的典型标记。

在 LED 照明下未观察到这种转变(图 3,右),表明潜在电子跃迁的吸收截面较小和/或量子产率较低。因此,拉曼探测激光器的高光子辐照度|| 是进行光反应所必需的。根据红外数据(图 S3 †和3D ,右),等式 (1)的氧化还原平衡通常朝向左侧,这意味着 [FeFe] 部分的还原电位高于 [4Fe4S ] 子站点。
从热力学的角度来看,这种平衡预计不会在低温下逆转,表明 H' red在这些条件下通过阻碍逆反应在动力学上稳定。

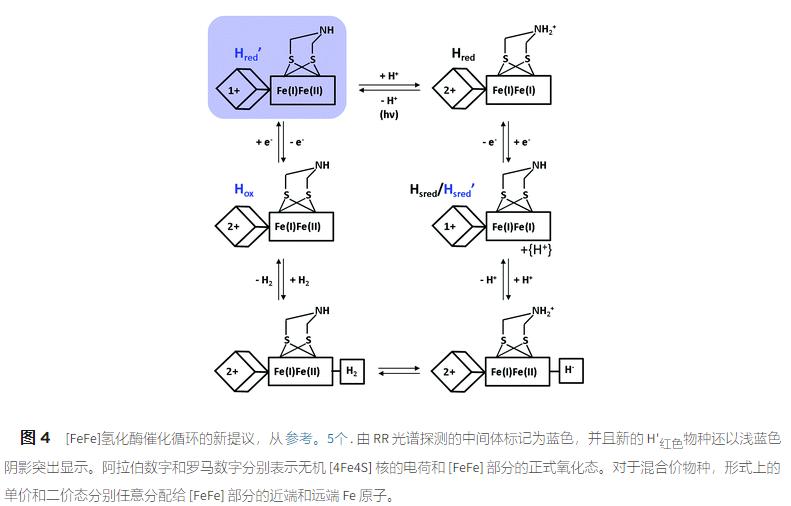
尽管在 RR 实验中对 H'红色物质 的富集有特殊要求,但这种中间体可能对催化循环有合理的贡献(图 4)。值得注意的是,之前已经提出了根据等式 (1) 的热平衡,但在本研究之前,尚未通过实验观察到右侧配置 H'红色。
根据我们的发现,我们提出该物种代表了氢析出过程中的第一个 H 团簇中间体,它是由 H ox的单电子还原形成的,并通过分子内电子和质子转移到 [FeFe]快速转化为 H red子站点(图 4)。
四、结论
总之,这项工作提供了对整个 H 簇及其辅因子构建块的 RR 可检测金属-配体模式的详细见解。使用这些振动标记作为 [FeFe] 和 [4Fe4S] 子位点氧化还原态的敏感探针,发现了H 团簇的新型中间体 H' red 。
根据来自非天然 H 簇衍生物的数据,这种亚稳态物种被认为具有不寻常的 [Fe I Fe II ]、[4Fe4S] 1+基态和去质子化的 adt 配体,可识别 H'红色作为 [FeFe] 氢化酶生物制氢过程中缺失的第一个 H 簇中间体。 这一发现代表首次检测到在稳态条件下无法获得的这些酶的瞬时中间体。从更广泛的意义上讲,本研究强调了低温光谱学探测和表征氢化酶和相关金属酶中其他无法获得的中间体的能力。

参考文献:
1、JW Peters 、 WN Lanzilotta 、 BJ Lemon 和 LC Seefeldt , 《科学》,1998 年, 282 年 ,1853 年
2、Y. Nicolet 、 C. Piras 、 P. Legrand 、 CE Hatchikian 和 JC Fontecilla-Camps , 结构,1999 年
3、W. Lubitz 、 H. Ogata 、 O. Rüdiger 和 E. Reijerse , 化学。牧师, 2014,
4、TG Spiro 和 RS Czernuszewicz , 酶学方法。1995,
5、MK Akhtar 和 PR Jones , Appl。微生物学。生物技术。2008,