这篇分享的灵感来源是王晓东院士课题组发表的molecular cell文章。看完很有感触,每次参观大牛实验室,墙上大部分都是CNS的文章,有一部分会是molecular cell文章,老点的会是JBC。拿我某师兄一句话,大牛的工作都是冲着cell去的,等做完了,如果意义不大,就投投molecular cell,如果还是不行,JBC算了。这三个杂志有个共同点,在机制上做得非常严谨。
今天分享王院长课题组的2篇文章:2009年的一篇cell,2012年的一篇cell。文章中的科学意义就不细说了,主要看思路,以方便我们自己在研究时参考。
壹
Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. CELL. 2009.
01
细胞模型及表型(检测指标)
根据文章的描述,在研究细胞凋亡时发现,凋亡诱导分子Smac模拟物可以诱导很多细胞凋亡,且这个凋亡可以被caspase的*制剂抑**z-VAD所抑制,很好的结果。但是,在结肠癌细胞HT-29中,Smac模拟物可以诱导细胞death,但这种death不能被z-VAD所抑制,从而提出在HT-29中Smac模拟物可通过necrosis(坏死)路径促进细胞death(死亡)。(这里, 一个是提出了细胞表型,一个是有了好的细胞模型 ,如果没有HT-29细胞坏死模型,这篇文章就不能继续了。 论细胞模型的重要性。 )
02
功能分子筛选及验证
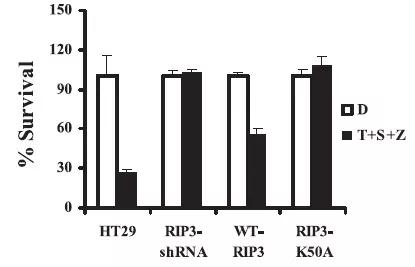
有了很好的表型,就开始筛选功能基因了。这篇文章用人的22000个siRNA的化学文库进行了筛选( 我们现在是用的慢病毒介导的siRNA文库,对细胞的感染效率更高,也更稳定,HCS功能筛选。 )发现:在坏死诱导建模下(T+S+Z),RIP3干扰后细胞存活增加。(T为TNF-α,代表炎症诱导的坏死;S为Smac模拟物,诱导坏死;Z为z-VAD,抑制凋亡只看坏死。)
RIP3是激酶,那么其功能是否与其激酶活性相关呢。构建了激酶失活的突变体(K50A),发现,激酶活性丢失后,其促坏死的功能丧失。( 激酶研究,探讨其激酶活性与功能的关系,体现工作严谨的地方 。)
这个实验其实也是 siRNA的回复实验 ,第三组和第四组都是在RIP3-干扰的前提下进行的。第三组是把RIP3不能被干扰的野生型转进去,看对功能的回复,有回复作用。第四组是把不能被干扰的RIP3激酶突变体转进去,发现没有功能回复作用。很完美的数据。
另外一点,这里如果不做第三组的回复实验,就需要展示RIP3两条有效片段的数据,以说明第二组的结果,不是由脱靶造成。高分文章的严谨性。
再有一点,文章中用的是RIP3-shRNA的稳定株,这里要注意,因为RIP3干扰后,细胞活得好好的,可以建干扰稳定株。但也需要尽快用,不然可能会有干扰效果丢失。如果基因干扰后会影响细胞的存活,那么就不建议筛稳定株了。
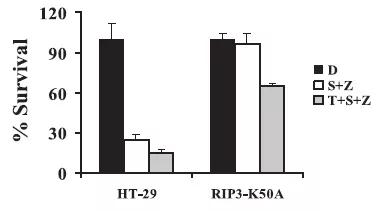
如果不能建干扰的稳定株,其实还可以做过表达。这篇文章也做了。把激酶突变体构建稳定株,然后看对功能的影响。发现突变体明显要弱于对照细胞株(严谨来说,其实对照应该用野生型的稳定株)。(做实验的时候,如果一个路径不行,再试试另外的路径)
03
动物实验
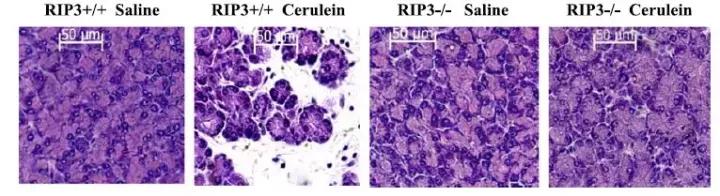
因为RIP3是激酶,激酶是很好的候选药靶,在功能明确的情况下,值得花经费构建转基因动物。这篇文章构建了RIP3敲除动物。前面研究是在TNF-α刺激情况下造成细胞坏死。因此在敲除动物上诱导了胰腺炎的模型(Cerulein为诱导剂),然后看组织损伤情况。 从基础研究到疾病。
RIP3敲除后,胰腺炎中,组织损伤减少。从临床转化的角度,筛选RIP3激酶*制剂抑**,可能为胰腺炎治疗的先导化合物。
贰
Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. CELL. 2012.
第一篇文章中RIP3是激酶,按照研究的思路机制上应该找其发挥功能的底物是谁。2009年没有找吗?不知道,反正RIP3的底物在2012年又发了一篇cell。(提醒,2009年的文章中不是没有机制,是做了RIP3的上游。)
01
细胞模型及药物筛选
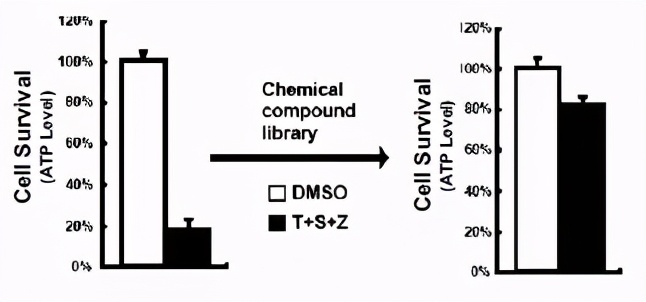
从临床转化的角度,2009年发现了很好的候选药靶,应该可以筛选先导化合物了。采用了前面使用的细胞建模条件,进行了200000个化合物库的筛选,找到了一个化合物可以抑制HT-29在建模条件下的死亡。后续对此化合物进行了修饰后获得了更好的*制剂抑**Necrosulfonamide(NSA)。
一般来说,有了激酶,就筛选激酶的*制剂抑**,但是不同的激酶,活性域结构都差不多, 通过激酶活性筛选出来的*制剂抑**,往往不特异 ,如果上临床,副作用会大。不知道王院士是否是考虑到这一点,从而没有直接筛选激酶的*制剂抑**。
通过细胞活性筛选的*制剂抑**,不一定跟RIP3相关,因此文章中探讨了*制剂抑**与RIP3的关系。证明*制剂抑**与RIP3相关。
02
激酶底物筛选
从研究的角度,有了激酶,需要找到其发挥作用的磷酸化底物。文章中用RIP3进行IP-MS,找到了MLKL。其实,这里只能说明找到了RIP3直接互作的一个蛋白MLKL,MLKL是否受RIP3磷酸化,不知道。其是否有促坏死的功能,也不知道。
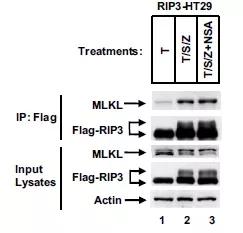
03
底物功能
对筛选到的互作蛋白MLKL进行功能探讨。发现,MLKL干扰后,细胞活得多了。( 机制研究,功能相关性机制 。不清楚的,听场季博的讲座。)
注意,下面这个功能实验,也做了功能回复实验。以证明干扰靶点没有脱靶。
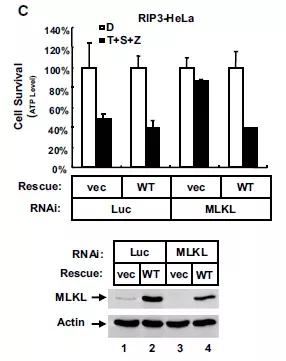
04
药物与靶点关系验证
把NSA偶联生物素,然后去拉蛋白。这里注意,NSA偶联生物素后需要检测是否活性还在。 活性还在的情况下再去拉蛋白 。结果显示,拉下来的蛋白中有MLKL。
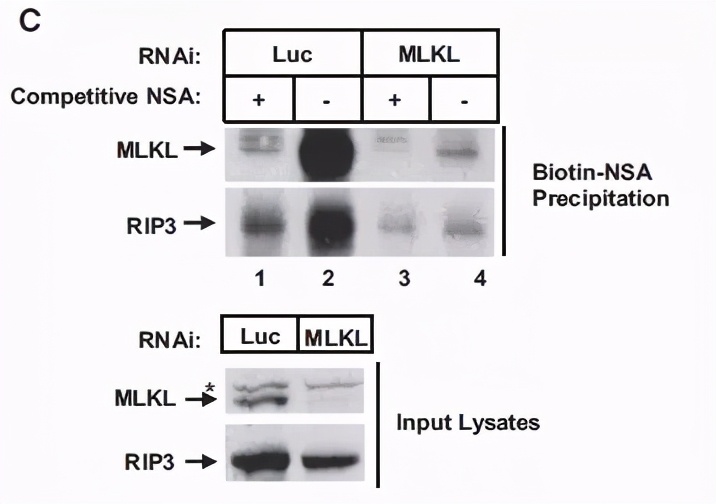
且发现NSA与MLKL的结合是共价结合。把MLKL表达出来后,跟Biotin-NSA孵育,然后电泳,发现在MLKL条带处,能够检测到biotin。(*制剂抑**与靶点共价结合,对于临床转化来说,由于其与靶点结合后就不分开了,可能副作用会大。)
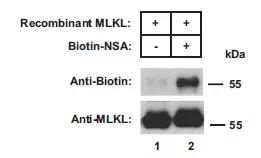
05
激酶与底物磷酸化关系验证
前面用RIP3拉到MLKL,只是证明有直接互作关系,不能证明MLKL是RIP3的底物。因此这一步就检测RIP3对MLKL的磷酸化,发现,在体外磷酸化实验中,RIP3可以磷酸化MLKL。到这里,才证明MLKL是RIP3的底物。
并通过质谱鉴定到T357及S358是RIP3磷酸化MLKL的位点。
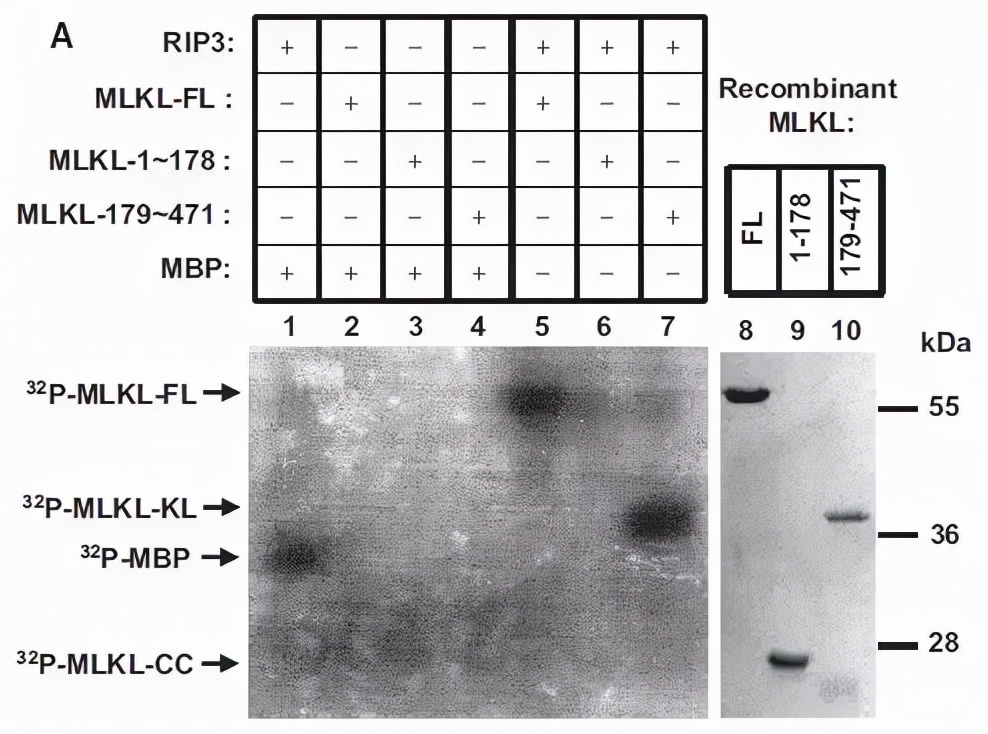
从研究的步骤来说,如何找到RIP3的底物,并拿到磷酸化位点呢。建议步骤是,a、激酶进行IP-MS,找互作蛋白,listA。2、激酶干预后进行磷酸化蛋白组检测,找磷酸化改变的蛋白,listB。ListA与listB的交集中去验证。交集的蛋白,有激酶互作,通过激酶干预后磷酸化也有改变。同时,磷酸化蛋白组的结果中也告诉我们改变的磷酸化位点是什么。一举多得。( 研究激酶的老师,一定要参考,比一步一步做要靠谱。 )
06
底物磷酸化位点功能验证
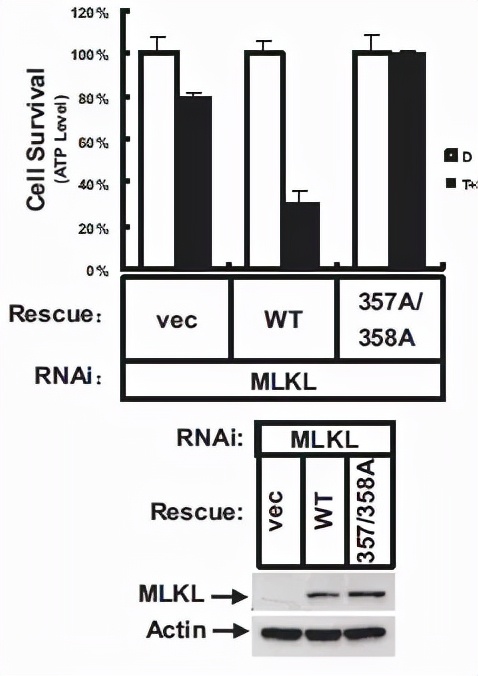
研究激酶与底物,底物磷酸化位点找到后,一定要证明这个位点的磷酸化与功能相关。因此就做了这两个位点的组成失活型突变体(T357A、S358A),发现组成失活型突变体不能促进细胞的坏死。
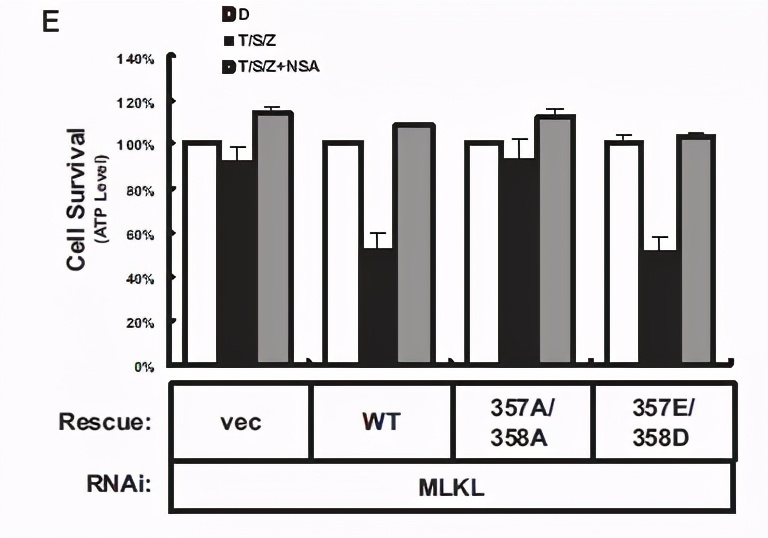
磷酸化位点,除了做组成失活型外,还可以做组成激活型。文章中也做了组成激活型(T357E、S358D),发现组成激活型与野生型功能一致。
文章中还做了很多个性化的数据,有兴趣的老师可以看看。
总结
激酶与底物的关系探讨工作,可以做得很细,很严谨。是高档次文章喜欢的内容。同时,激酶容易筛选*制剂抑**,是很好的候选药靶,临床转化喜欢的内容。
1、研究中,组学做完后,看组学中改变的激酶有哪些。
2、这些改变的激酶优先做功能探讨。可以跟第一篇文章学,采用HCS的方法找有功能的激酶。
3、找到有功能的激酶后,探讨其功能与其激酶活性是否相关。(活性位点进行突变)
4、验证出其激酶活性与功能相关后,找底物。(激酶IP-MS找互作蛋白,激酶干预做磷酸化蛋白组,两者取交集后验证。)(激酶做IP-MS,由于激酶结合底物后很快磷酸化并解离,这个IP-MS的数据不一定理想。而激酶失活突变体做IP-MS,结合的底物不能磷酸化,不能很快解离,从而容易检测到。但有些激酶需要自身磷酸化后才结合底物,这种情况下做失活突变体的IP-MS,还是拉不到底物。此步骤遇到问题的老师,找季博。)
5、底物进行功能验证,以证明底物功能与激酶一致。(交集比较多时,可以将这些底物拿功能HCS进行筛选)
6、拿到功能一致的底物后,进行激酶与底物进行直接互作验证。
7、从磷酸化蛋白组中找底物磷酸化位点,底物磷酸化位点进行功能验证。以证明底物发挥功能需要此磷酸化位点。(组成失活型突变为A,组成激活型突变为D或E)
8、根据研究的疾病及功能表型,补充其他细胞学实验。如果数据好,尝试构建转基因动物。
以上是激酶研究的思路,比较粗,每个课题都需要根据实际情况来进行调整。供大家参考。