理论计算在表面研究中的地位已经越来越重要了,但是随着计算工作的增多,水平层次也出现了巨大的分化。
为什么有的课题组可以每年在JACS上发表数篇文章?这些文章计算经验肯定是需要常年的研究经验积累的,也是作者的秘密。只要有人愿意诚心诚意的教,这些计算技巧和思路都是可以掌握的。
正在学习理论计算催化的同学,可以对比看一看,自己还有那些知识没有掌握?
一、程序编译
一些重要的计算功能原版的程序是没有的,必须编译插件,比如:
-
计算过渡态必须的VTST
-
优化二维材料必须的OPTCELL
-
溶剂化模型VASPSOL
-
计算化学键NBO/AdNDP
-
Wannier90,BEEF泛函等
二、复杂表面模型建模
-
构筑表面不同的terminal
-
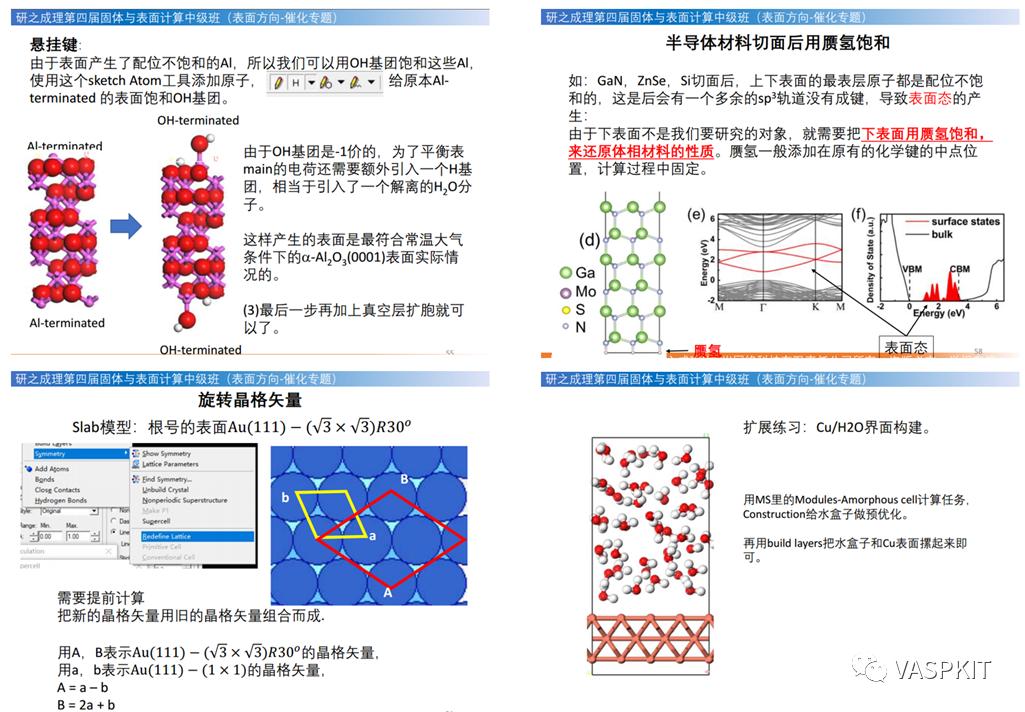
悬挂键/饱和赝氢
-
晶格矢量的旋转与根号表面wood标记法
-
异质结建模
-
负载催化剂团簇模型
-
优化模型显示效果
三、输入文件的处理
-
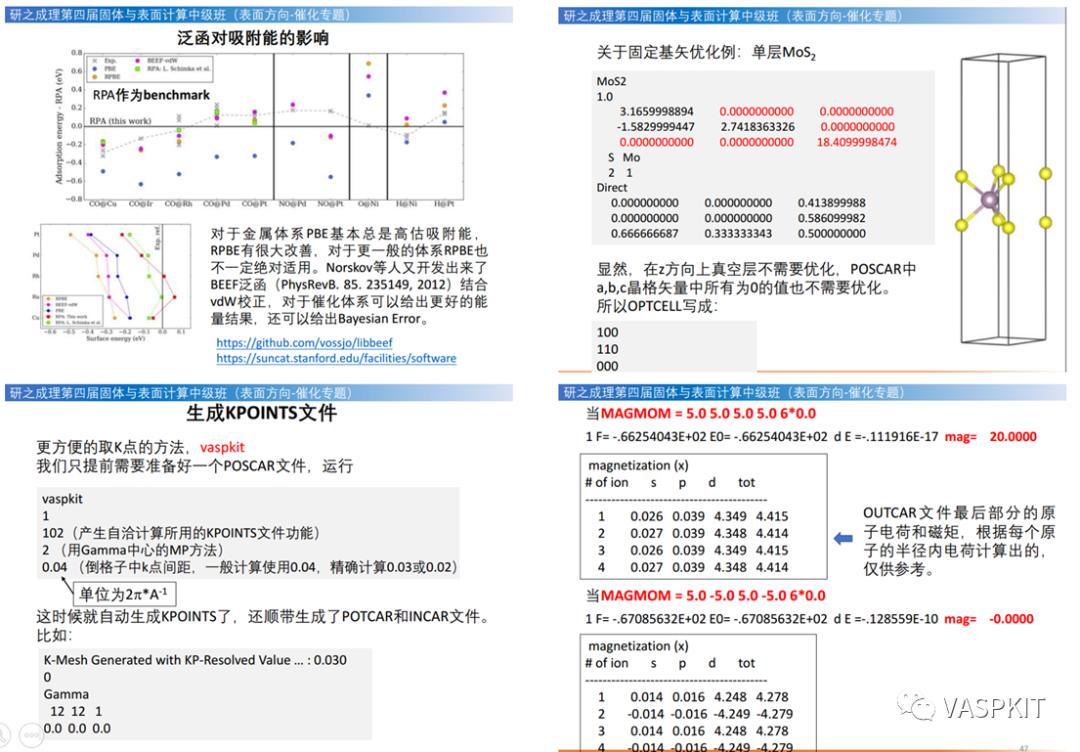
POSCAR技术:一键固定原子(按照层数和厚度),给同种元素定义不同赝势(赝氢),对9个晶格参数差异化固定(选择行优化晶格矢量)。
-
POTCAR技术:如何一键生成,自定义元素价电子数,修改元素质量(同位素模拟),不同core大小的赝势选择,自动按照vasp手册建议的赝势生成。
-
KPOINTS技术:按照倒空间密度一键生成K点文件。
-
INCAR技术:套模板一键生成计算输入文件。杂化泛函,范德华校正与相关泛函的设定方法,泛函对催化吸附能的影响。DFT+U设定,磁性体系的计算技巧,KPAR/NCORE/NPAR的设定技巧。
四、态密度DOS的计算分析
-
能带与态密度形成原理,超胞与能带反折叠,Peierls畸变与Jahn-Teller效应对能带态密度的影响。
-
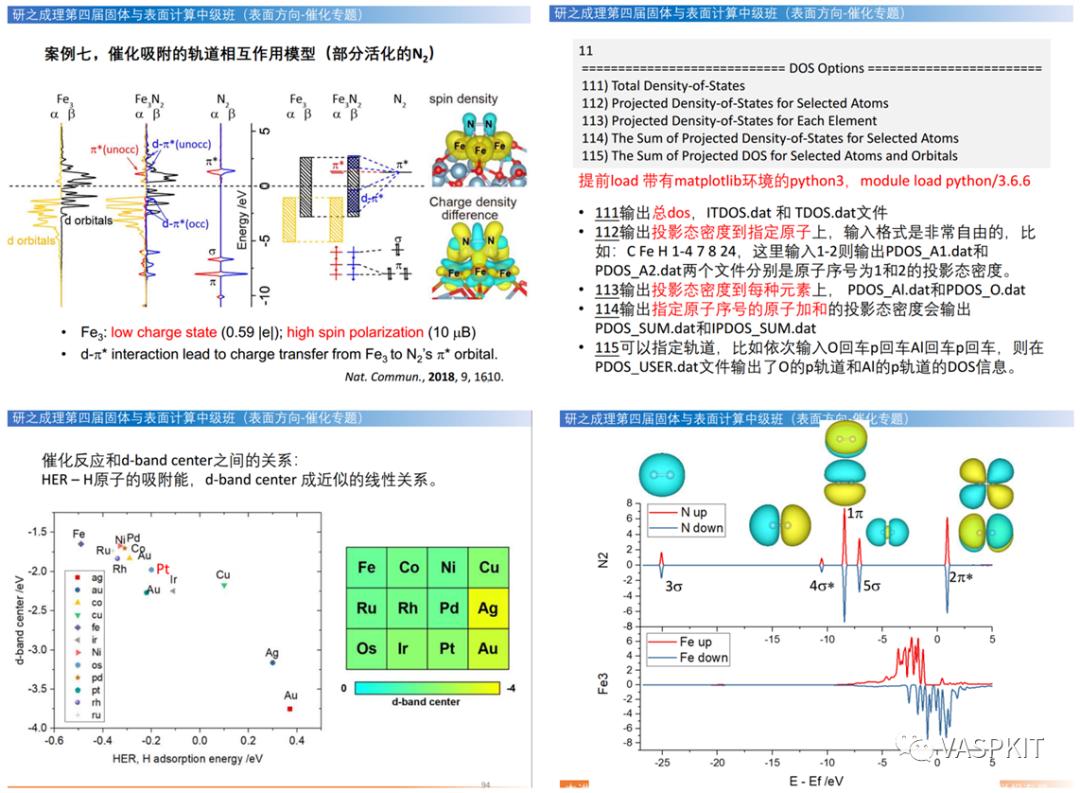
用DOS分析离子键和共价键,电子转移分析,表面的氧化还原特性分析,研究表面缺陷态与表面态,悬挂键对态密度影响,吸附分子和载体的相互作用模型分析。
-
P4VASP和VASPKIT后处理DOS方法,态密度的积分,成键和反键轨道分析。CO,N2,CH4等与表面相互作用的轨道分析方法。
-
d-band center理论与调控,一键计算d-band center。
五、电子结构分析
CHGCAR提取电荷密度、自旋电荷密度、调整格点密度、Bader电荷计算、原子电荷着*图色**、电荷密度差、变形电荷密度、实空间波函数、Partial charge density、STM模拟、静电势计算、功函数、分析异质结静电势与电子流向、分子表面的静电势分布、电子定域化函数。

六、表面热力学与动力学
表面吸附:化学吸附、物理吸附、解离吸附、吸附位点的综合考虑。吸附物种间的相互作用,覆盖度与温度、压力、吸附能关系,催化剂毒化机理。
有限位移法/DFPT法振动频率计算、零点振动能、振动方向可视化。热力学量的计算、引入反应温度、压力、pH、电极电势等等外界因素对自由能的影响、气体分子、吸附分子、固体的自由能校正,vaspkit热容、熵、自由能校正计算。
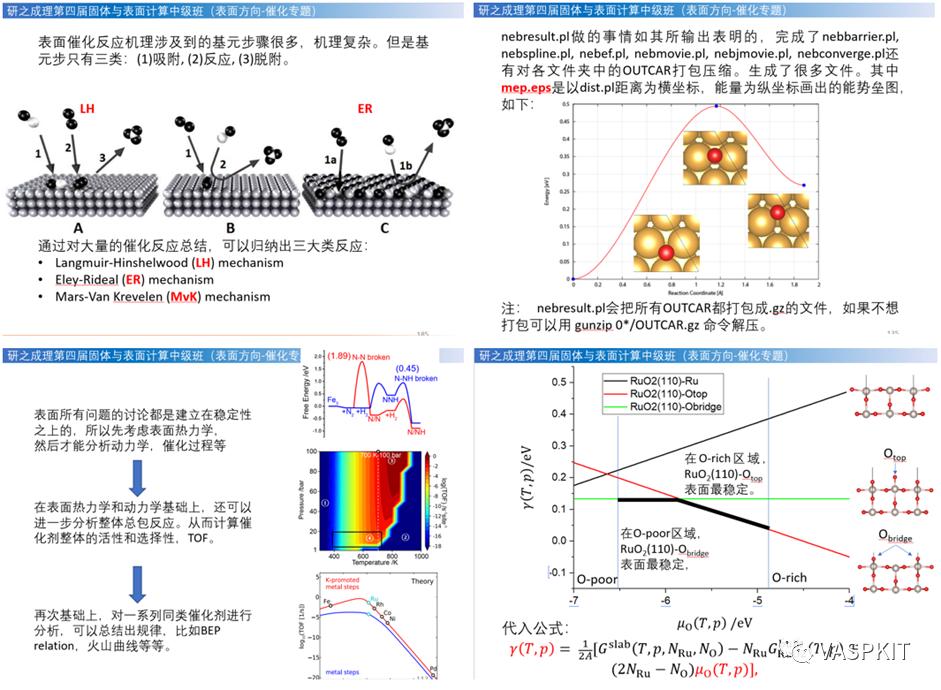
热力学分析:表面吸附平衡,熵变对吸附的影响,表面能计算,非化学计量比表面的热力学稳定性分析。化学势热力学相图绘制,温度、压力、吸附物对化学势影响。
过渡态搜索:过渡态判据,无过渡态反应类型,寻找过渡态方法综述,chain-of-states 搜索方法和原理,传统 PEB 方法(plainelastic band)方法遇到的问题,NEB 中 nudge 过程原理,Climbing Image 原理,判断插点个数,插点方法,nebmovie.pl 生成 movie 判断插点合理性,CINEB 的 INCAR 关键词和计算方法,VTST 优化算法解析,nebresults.pl,nebbarrier.pl, nebspline.pl, nebef.pl 等 VTST脚本,neb.dat、spline.dat、exts.dat、mep.eps 文件自动判断NEB路径上的极值点,Dimer 方法原理,选择 Dimer,平移 Dimer 过程,Dimer发展历史,Dimer 的 INCAR 关键词和计算方法, MODECAR 生成,DIMCAR、CENTCAR和 NEWMODECAR 解读,生成 DIMER 方向 movie 预判过渡态计算是否成功,VTST 过渡态计算收敛判断,过渡态搜索常见问题和解决方案。扩散系数与离子电导计算。NEB+Dimer 高效搜索过渡态,neb2dim.pl。NEB 计算线性插点的问题,IDPP 插点原理,使用 pymatgen-diffusion 和 pymatgen 做 IDPP 非线性插点。
化学反应速率常数计算:基元反应,单分子多分子基元反应速率方程,稳态平衡和速率方程关系,反应速率常数计算,Arrhenius equation,eyring equation 推导,指前因子计算,表观活化能拟合,过渡态理论,准平衡态假设,过渡态络合物,透热系数,Recrossing 问题,判断基元反应是否容易发生,Eyring 过渡态理论的局限性,ΔGa/温度/反应速率常数 k 之间的关系,半衰期,谐振过渡态理论 HTST 公式,吸附和脱附步骤反应速率,Knudsen-Langmuirequation,隧穿效应,Wigner 方法计算透热系数。
催化反应机理:Langmuir-Hinshelwood (LH) mechanism,Eley-Rideal(ER) mechanism,Mars-Van Krevelen (MvK) mechanism,及其速率方程组。LH 实例合成氨反应,ER 实例Volmer-HeyrovskyHER,MvK 实例低温 CO 氧化反应。催化循环和总包反应能量。
七、计算电催化
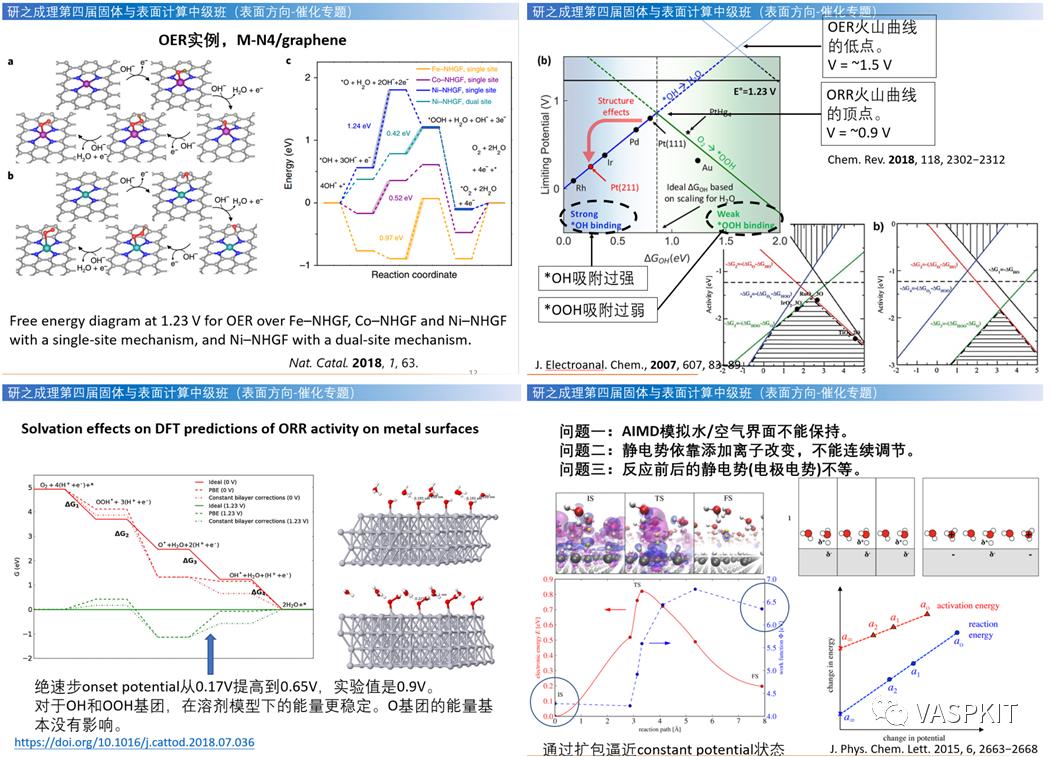
电催化相关简介,Norskov的电催化台阶图OER,ORR计算原理,实操Co-NC材料电催化台阶图的计算。酸性和碱性计算区别。HER,NRR,CO2RR计算原理,溶剂效应与双电层理论,显示/隐式溶剂模型理论,vaspsol基本原理。vaspsol的使用,计算溶剂效应对H2O表面电荷影响,GaN表面的溶剂化能。电催化火山形曲线的计算和分析。隐式溶剂模型和显示溶剂模型的使用。溶剂化效应对电催化计算得影响。恒电势计算与恒电荷计算方法。
课程内部交流群里有上万条答疑记录,永久答疑,相互学习,是学员共同进步的乐园。

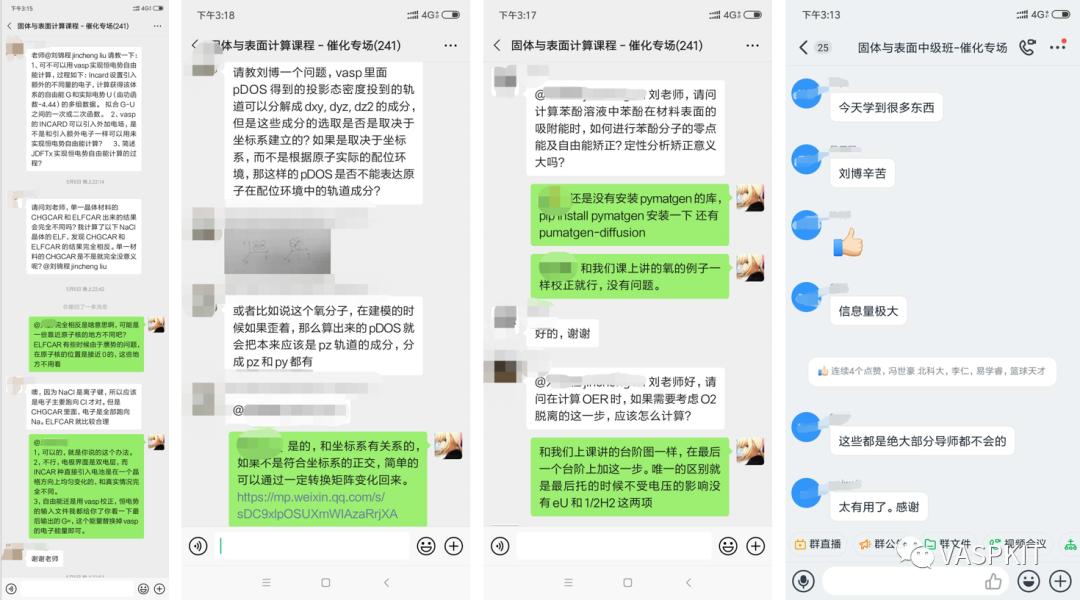
上车计算催化课的机会,以后永久回看专题课。加入全网最专业的计算催化学习圈子。享受课程视频永久回看,答疑微信群永不解散,另有500张PPT,近千个计算文件可供学习。答疑群已经有上万条的答疑聊天记录。
入圈价格:圈子价格1000元(自费),1200元(*票开**)。一次付费永久入圈。报名之后可以获得4天的计算催化课程视频,ppt,计算文件,加入答疑群和催化计算学习圈子。报名添加客服微信: yansci。
注意:此课程不是0基础的课,如果完全没有计算经验的同学,需要根据自身情况先加入qvasp与第一性原理计算圈子学习。
讲师介绍:
刘博士,清华大学博士,长期从事表面催化和材料计算研究,对量化计算,第一性原理计算,分子动力学模拟有六年研究经验,精通 VASP,CP2K,Gaussian,ORCA 熟练使用 JDFTx,Gromacs,ADF 等计算程序。近三年在表面催化,电催化,电池材料,过渡金属配合物等领域发表多篇文章。在 JACS,Nat. Commun. ,ACSCatal.,Nano Energy, Natl. Sci. Rev. 等期刊上以第一作者发表纯理论计算文章,并与实验科学家合作在 Nat. Nanotech., Nat. Commun., PNAS,Adv.Mater., JACS 等期刊上发表多篇合作文章。
Google Scholar:
https://scholar.google.com/citations?user=hAkSR2wAAAAJ&hl=en
助教老师:
唐博士,比利时列日大学博士后。目前是多个理论计算平台(学术之友微信公众号、DFT 计算之家QQ 群等)的运营者,关注粉丝量超过万人。主要从事偏固体物理方向的理论计算。其中第一作者论文发表在 J. Phys. Chem. Lett.,J. Mater. Chem. C 等国际期刊上,具有多次线下软件安装和计算培训的经验,培训学员超过 100 人。
未来计划的专题讨论课:
-
COHP计算化学键强度(此专题同步在催化中级班课程群直播)
-
周期性体系的NBO和SSAdNDP计算
-
单原子催化剂的理论计算
-
高通量计算与Pymatgen自动化处理表面结果
-
光催化计算与band alignment
-
ASE辅助工具的使用
-
JDFTx恒电势方法电化学计算
-
电子化合物的计算
-
显示溶剂化模型与电催化过渡态
-
异质结模型的程序化构建
-
催化体系微观动力学分析
-
CATMAP计算催化动力学