

今天是2月的最后一天,也是国际罕见病日,今年的主题是“研究”,口号是:“研究带来无限可能”。
相信不少人会心生疑问,罕见病离我很遥远,生活中几乎很难碰到这样的例子。其实,之前风靡一时的“冰桶挑战”其实就是针对肌萎缩侧索硬化(ALS)这个罕见病的一项公益活动,今天,健康君就给大家介绍几种儿童罕见病,也许他们离你很远,但愿我们更多了解、关注,能帮助他们摆脱孤独。

罕见病受害者多数为儿童
咱先从罕见病说起吧~究竟啥是罕见病呢?
罕见病又叫“孤儿病”,根据世界卫生组织(WHO)的定义,患病人数占总人口数的0.65‰~1‰的疾病统称为罕见病,目前经确认的罕见病近7000种,约有80%的罕见病是由遗传缺陷所致。复旦大学出生缺陷研究中心的数据显示,我国罕见病群体人数约1680万人。
对于那些被罕见病折磨的患者们来说,他们的病症通常都是一种孤立的存在。对于病症的整体知识很少,诊断和治疗的过程往往是长时间而且非常痛苦的。这些罕见病受害者大多数是年轻人或是儿童。
广州市妇女儿童医疗中心遗传与内分泌科主任、广东省医学会罕见病学分会主任委员、主任医师刘丽表示,有70%—80%的罕见病会在儿童期发病,大多数罕见病严重的会致死致残……
那么除了我们听过的渐冻人症,还有哪些儿童罕见病呢?
1
成骨不全症
成骨不全又称脆骨症,也有称“瓷娃娃”,主要特征有骨质脆弱、蓝巩膜、耳聋、关节松弛,是一种由于间充质组织发育不全,胶原形成障碍而造成的先天性遗传性疼痛。这种疾病即使是轻微损伤,也可能引发骨折,如果病情严重,还会自发性骨折,而且没有特殊的治疗方法。

2
法布雷病
法布雷病(Fabry病)是一种十分罕见的X染色体连锁遗传的鞘糖脂类代谢疾病,这是一种被称为人体细胞溶酶体脂蛋白代谢异常的疾病,因为身体里缺少一种酶,导致有些脂蛋白无法分解,堆积造成各个系统病变。比如,它可造成四肢非常剧烈的疼痛,并对肾、心脏、脑、神经等各器官产生严重损害造成病变,病情呈进行性加重发展态势,如得不到有效治疗将危及生命。

目前法布雷病只有美国健赞、爱尔兰Shire、韩国绿十字、美国Amicus Therapeutics生产的药物可治,而前三家公司的药物都只能在18岁以后才能使用,均为静脉注射,但国内并没上市。这些药物使用价格高昂,如健赞公司的药大约200万元/年,且需终身使用,并非一般家庭所能承受。
3
Prader-Willi综合征
Prader-Willi综合征,又称Prader-Labhar-Willi综合征、隐睾-侏儒-肥胖-智力低下综合征、肌张力减退-智力减退-性腺功能减退与肥胖综合征,是一种与基因组印迹相关的遗传性疾病,新生儿发病率为两万分之一。1965年,该病由Prader等首次报道,至今报道的病例仅数百例。
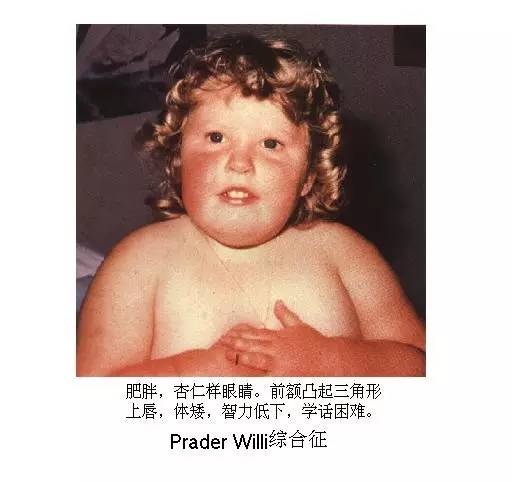
光看这长长的别称,健康君就懵了……2015年,首儿所呼吸内科曾接诊过一位4岁患者,其主要的症状是肺炎、过度肥胖、生长发育迟缓、智力发育不良等,后经腺样体、扁桃体切除手术后,肺炎和呼吸暂停的问题解决了,但这位小患者却要长期打生长激素,并进行相关检测和评估……
4
朗格罕细胞组织细胞增生症
朗格罕细胞组织细胞增生症(Langerhans cell histiocytosis,LCH),是一组来源于骨髓的朗格罕细胞的异常增生,伴有数量不等的中性粒细胞、嗜酸性粒细胞、淋巴细胞、浆细胞及多核巨细胞浸润,引起组织破坏的疾患。
LCH 在任何年龄均可发病,但低龄儿童发病率较高,高峰年龄为1-3岁,15 岁以内儿童发病率为0.02-0.2/万,男女之比约为1.6-2:1。这种病临床表现差异较大:
可能长期的表现仅仅是轻微皮疹,单部位溶骨性损害,没有明确的临床进展。
也可能表现为多个重要脏器受累,病情进展迅速,甚至导致死亡。
有报道称,此病最常累及的脏器有骨骼、皮肤和垂体等,超过50%的病人在初始治疗后,出现难治或复发,且大多数难治复发病人多发生在最初2年。
5
反复湿疹血小板减少免疫缺陷综合症
啥?湿疹也列入罕见病了吗?非也!
反复湿疹伴血小板减少免疫缺陷综合症,又名Wiskott-Aldrich综合征(WAS),是一种X-连锁的原发性免疫缺陷病,以免疫缺陷、湿疹和血小板减少三联征为典型临床表现,不典型者主要表现为血小板减少,而无明显免疫缺陷表现,此时需与特发性血小板减少性紫癜鉴别。
此病血液系统表现常较突出,生病后即可发生出血倾向,包括紫癜、黑便、血尿等,血小板明显减少,血小板体积变小。湿疹血小板减少伴免疫缺陷综合征的发病率为十万分之一。
6
黏多糖贮积症
黏多糖贮积症是一组因降解各种黏多糖相关的溶酶体酶先天性缺陷,使不同的黏多糖不能完全被降解,而在各种组织溶酶体和细胞外基质内沉积,导致组织和器官的功能损害。临床上MPS分为Ⅰ型、Ⅱ型、Ⅲ型、Ⅳ型、Ⅵ型、Ⅶ型及Ⅸ型等。
主要影响的器官有:心脏、肺脏、骨骼、关节、胃肠系统和中枢神经系统。研究表明黏多糖贮积症的群体发病率约为1/40万。
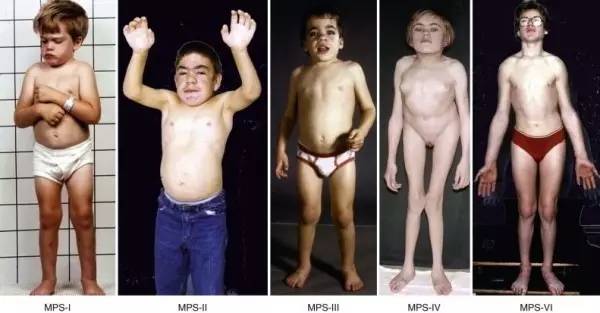
目前,黏多糖贮积症1型的治疗药物已于2005年在美国上市,但在中国却并未上市。
7
Alagille综合征
Alagille综合征是一种可影响肝脏、心脏、骨骼、眼睛和颜面等多系统或器官的显性遗传性疾病,常以婴儿期胆汁淤积为突出表现。该综合征在1969年由Alagille等首次报道,国外报道该病的发病率约为1/70000。国内近年来也开始关注此病。
8
永存第五弓
永存第五弓是一种罕见的先天性心脏病,自1969年Van Praagh首次描述这种畸形以来,全世界仅有一些个案及小组病例陆续被报道。国内上海儿童医学中心曾报道5例手术,但手术死亡2例,死亡率高达40%。
9
先天性胫骨假关节
先天性胫骨假关节(CPT)是一种罕见的疾病,发病率为1/14-19万,其特征为胫骨节段性发育异常、无正常骨形成,伴随成角畸形、病理性骨折和骨不连接。由于发育异常所致胫骨的畸形和特殊类型的不愈合,最终形成局部的假关节。
此病病因复杂,跟染色体异常有关,常合并神经纤维瘤病和皮肤咖啡斑,手术治疗困难,多数要截取病侧肢体安假肢度过一生。
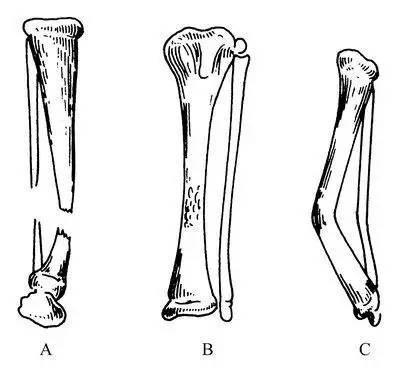
这么多种不同的罕见病,背后却是数百万儿童难以治愈的一生……
据不完全统计,每年新出生罕见病患者超过20万,但得到明确诊断的不足40%。罕见病患者的确诊时间平均需要5年,平均诊断成本大于5万,如果一个患者不能得到及时诊断,就更谈不上有效的治疗。罕见病种类繁多,大多数缺乏药物治疗。有药用不上和无药可用,对罕见病患者来说都是一种不幸。
虽然有些罕见病已有治疗药物,但在国内却并未上市,罕见病发展中心(CORD)主任黄如方及健康君曾采访过的多位罕见病领域专家、患者均认为,国家医保政策应保障罕见病群体,将价格高昂的罕见病用药纳入医保体系,让治疗可及。
健康君也希望,罕见病的治疗能够获得更多关注,让“孤儿病”的患者不再是病患中的孤独者。
部分内容来自 首都儿科研究所
采写 张秀兰 王卡拉
编辑 火星Lu