
EGF和EGFR的发现
说到EGF/EGFR,不得不提到一位美国的生物化学家和诺贝尔奖获得者——斯坦利·科恩(Stanley Cohen)。1953年作为圣路易斯华盛顿大学的一名教员,他与意大利神经生物学家丽塔·莱维·蒙塔尔奇尼(Rita Levi-Montalcini)合作发现了神经生长因子(NGF, nerve growth factor)。

Stanley Cohen (1922–2020)
而表皮生长因子 (EGF, Epidermal growth factor)的发现是科恩在NGF研究工作中派生出的一个成果——一道充满惊喜和意外的附加题。
1959年,科恩作为生物化学助理教授来到范德堡大学,当他使用含有NGF的唾液腺提取物注射给新生小鼠后,发现了一个意想不到的“副效应”动物的眼睑提前睁开,牙齿也提前萌发,可是NGF纯品则没有这种作用。这提示着在提取物中肯定存在其它的生物活性因子,而这种因子同样具有生*刺长**激作用。1962年科恩分离出了这种因子,并取名表皮生长因子EGF。随后,在70年代初搞清楚了这种蛋白质的分子结构——由53个氨基酸残基组成的一种小肽。
在接下来的几十年里,科恩坚持不懈又卓有成效地探索了 EGF 的作用机制。在研究EGF及其它生长因子的过程中,最令人费解的谜题莫过于这些因子的调节信息是如何从细胞膜传入细胞核内的。科恩认为EGF应该先与细胞膜表面的受体结合,然后,EGF-受体复合物再转入细胞内产生生物效应。表皮生长因子受体EGFR(epidermal growth factor receptor)的鉴定为阐明表皮生长因子在靶细胞中激发细胞增殖等生物反应的机制提供了一个起点。

1986年的诺贝尔生理学医学奖
NGF与EGF的发现使得蒙塔尔奇尼和科恩两位科学家荣获了很多荣誉和奖项,最终,1986年的诺贝尔生理学医学奖也颁发给了他们。诺贝尔评选委员会认为“他们共同发现了控制细胞生长和发育的因子,这些发现为基础医学研究开辟了很多重要的领域,让人们对发育畸形、老年痴呆、经久不愈的伤口以及肿瘤都有了更多的理解”。
但是在诺贝尔委员会做出预见性的决定时,他们没有预料到的是科恩的EGFR 研究为开发用于抗肿瘤的靶向治疗奠定了基础,因为那时距离肿瘤精准医学的出现还有10多年。
EGFR及相关通路
科恩之外,对于EGFR 比较系统的研究开始于1977年,Falricat等筛选了多种细胞株,发现一些肿瘤细胞株含有很高的EGFR;1981年Shih等在脑细胞瘤中发现原癌基因NEU;随后研究者们又发现NEU基因,EGFR基因与禽类病毒致癌基因v-ErbB具有同源序列,所以把他们及之后发现的2个受体都归类为EGFR家族,并进行统一命名,分别为:EGFR (ErbB-1/HER1)、ErbB-2 (Neu,HER2)、ErbB-3 (HER3) 和 ErbB-4 (HER4)。这一家族都是受体酪氨酸激酶(receptor tyrosine kinase family,RTK),有高度同源的氨基酸序列及相似的结构特征,但配体各不相同。
第一个被发现的人表皮生长因子受体EGFR (ErbB-1/HER1)是其中最为广泛研究的,基因位于第7号染色体,由28个外显子组成,编码1186个氨基酸,糖蛋白分子量约为170 kDa,广泛分布于除血管组织外的上皮细胞膜上。EGFR与其它RTK有着一致的结构:含有配体结合位点的细胞外结构域、单次跨膜的疏水α螺旋区、以及含有酪氨酸蛋白激酶活性的细胞内结构域。基于这一结构,EGFR受体激活由一系列相应的事件触发:
-
配体结合:每个配体与受体的胞外结构域结合,EGFR的配体包括EGF,转化生长因子-α (TGF-α),双调蛋白 (AR), β 细胞素 (BTC),肝素结合表皮生长因子 (HB-EGF) 和表皮调节素 (EPR)
-
受体二聚化:配体与EGFR结合后,改变EGFR的三维构象,使EGFR形成同源二聚体或EGFR与ErbB家族另一成员结合形成异源二聚体
-
二聚化的受体发生交联磷酸化,即一个受体使另外一个受体上特定酪氨酸残基磷酸化,激活胞内区的酪氨酸激酶(TK 亚区),从而启动下游最主要的信号通路包括 JAK/STAT通路、P13K/AKT 通路、MAPK/ERK通路,PLCγ/PKC通路
通过这样的方式,有丝分裂信号从细胞外传递到细胞内,从而有效调节细胞的存活、生长、增殖、分化、血管生成、抗凋亡、迁移、侵袭、黏附和DNA损伤修复等等一系列的过程。
简单理解也就是说跨膜的EGFR蛋白一端伸出细胞膜外用来与生长因子结合,另一端则在细胞内连着蛋白激酶,一旦生长因子受体的一端与生长因子结合,另一端的激酶也会被激活并释放,使得蛋白磷酸化,然后一层层激活下游信号通路,触发一系列级联效应,来调控细胞的方方面面。
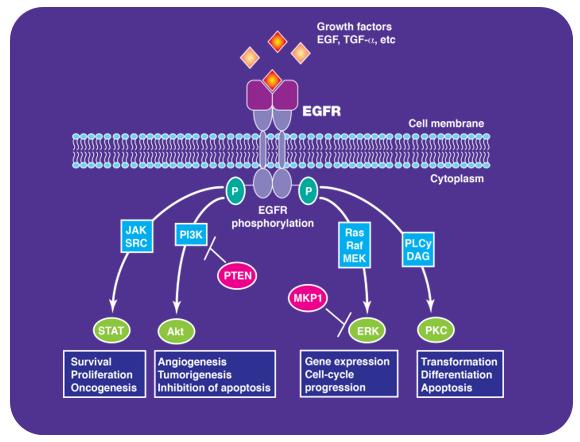
图片参考来源:Lee J, Moon C. Exp Biol Med 2011;236:375–389
EGFR信号通路与肿瘤的发生发展
肿瘤是一类复杂,多因素,毁灭性的疾病,多年来一直困扰着研究人员和临床医生。2000年和2011年由同一作者发表在《Cell》杂志上的2篇经典综述,hallmarks of cancer / hallmarks of cancer: the next generation,不断揭示区别于正常细胞的肿瘤的生物学特征,以及以此为基础的肿瘤治疗手段的研发。
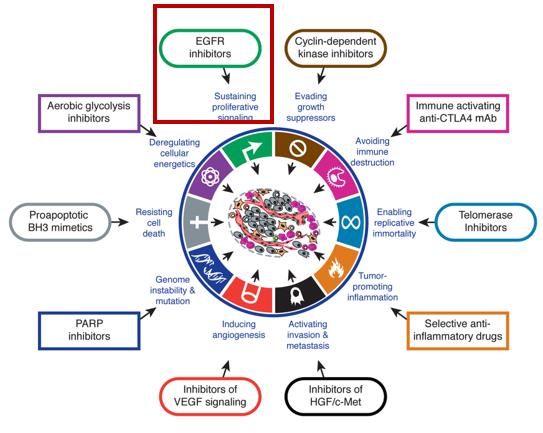
自给自足的生长信号是肿瘤非常关键的一个生物学特征。正常的细胞在从静息期变为活化增殖期是需要有丝分裂生长信号,这些信号通过EGFR这类的跨膜受体来传递给细胞,也就是说没有正常细胞是可以在不存在这些刺激信号的情况下去增殖的。但肿瘤细胞的行为却与此形成了鲜明的对比,肿瘤细胞对外源性生*刺长**激的依赖性显著降低,打破了正常细胞至关重要的稳态机制。
肿瘤细胞打破这种稳态的分子策略可以简单概况为以下三个方面:
-
改变胞外的生长信号水平:许多肿瘤细胞获得了合成并分泌自己生长因子的能力,于是它们可以相互激发并创建一个反馈回路,促使更多的肿瘤细胞分裂,同时也合成更多的生长因子
-
调节自身的生长因子受体:让细胞膜表面的生长因子受体远大于正常细胞,意味着肿瘤细胞对于肿瘤微环境中的生长因子十分敏感,对于正常细胞而言不足以触发细胞生长的生长因子水平在肿瘤细胞中却是有效的;此外,还可以让自身生长因子受体的结构发生改变,永远处于激活的状态,不再需要生长因子
-
肿瘤细胞可以改变信号转导的下游通路,生长因子和受体的调控就会被绕过
这些促进肿瘤发生发展的策略在不同瘤种或者不同的时期都各有体现,多种肿瘤类型中都观察到的EGFR过度表达就是其中的策略之一,并也因此启发靶向的抗肿瘤治疗。
不同瘤种EGFR高表达的情况
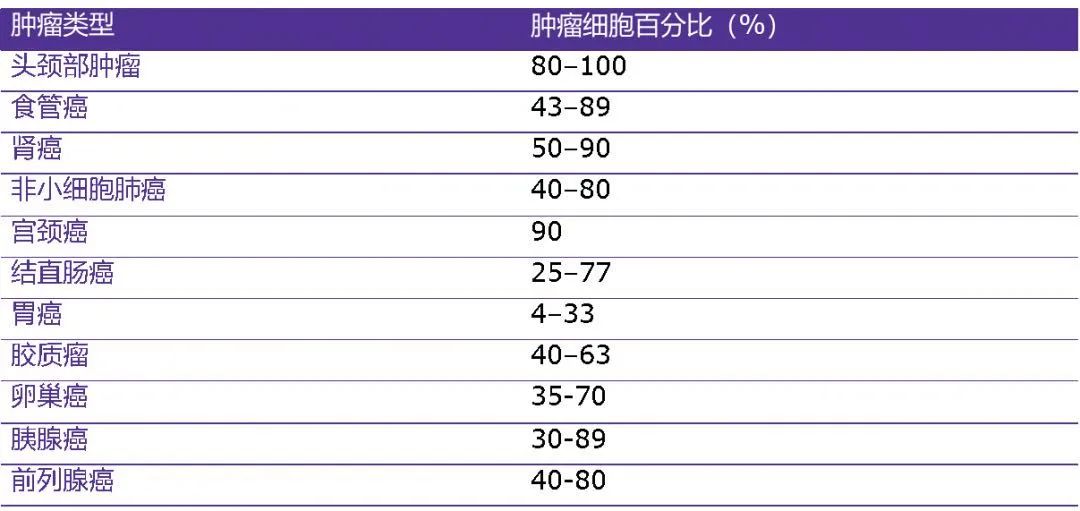
数据参考来源:Hanahan D, et al. Cell 2000;100:57-70; Hanahan D, et al. Cell 2011;144:646-74;Herbst RS, et al. Cancer 2002;94:1593–1611.
头颈部鳞癌的抗EGFR治疗探索和发展
EGFR的高表达可见于多种肿瘤类型,如各种上皮来源的肿瘤,尤其是鳞状上皮癌。例如在头颈部鳞癌SCCHN中EGFR高表达可见于80-100%的肿瘤细胞,这种过度表达的EGFR使下游信号通路不断强化,促进肿瘤细胞的体内外增殖,促进肿瘤新生血管的生成,促进肿瘤的远处转移,或者保护肿瘤细胞不进入凋亡等等。所以,通常EGFR蛋白高表达的患者其生存预后要更差。
目前进入临床实践靶向EGFR家族来治疗肿瘤的药物分为两类,抗EGFR单克隆抗体以及EGFR酪氨酸激酶*制剂抑**,在头颈部肿瘤中这两类药物也同样进行了探索:
01 抗EGFR的单克隆抗体
抗EGFR单克隆抗体通过识别受体的胞外区,与配体竞争性结合EGFR,干扰EGFR的自身磷酸化及阻碍细胞表面EGFR二聚体的形成,抑制信号转导通路的激活,从而抑制肿瘤细胞增殖,转移和血管形成。
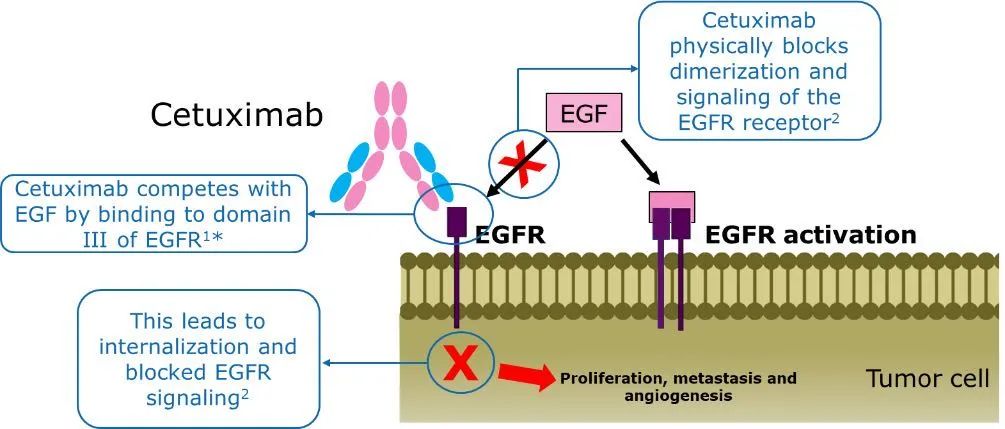
图片参考来源:Voigt M, et al. Neoplasia 2012;11:1023–1031; Adams GP, Weiner LM. Nature Biotechnol 2005;23:1147–1157.
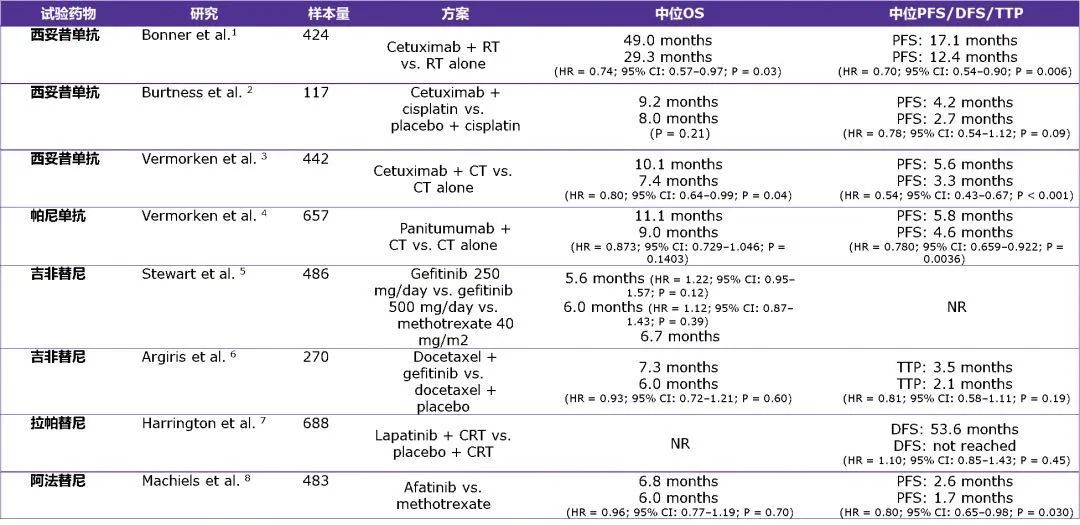
从肿瘤EGFR通路发现至今为止,西妥昔单抗(Cetuximab)仍然是第一个也是唯一一个在头颈部肿瘤中证实治疗有效性的单克隆抗体, 已经获FDA/EMA批准用于局部晚期以及复发转移SCCHN的治疗,2020年2月25日也获得中国国家药品监督管理局的批准。西妥昔单抗是IgG1型的人鼠嵌合抗体,该抗体最初源自一个小鼠骨髓瘤细胞系,其构建过程为将抗EGFR 的小鼠单克隆抗体(M225)的重链和轻链克隆,改造后与人κ-轻链和人γ-1重链的恒定区一起表达。这种嵌合的过程使得产生的抗体与EGFR 的亲和力比内源性配体例如EGF和TNF-α 高一个对数级,从而有效的胞内结构域的活化和后续酪氨酸激酶依赖的信号转导通路。同样,西妥昔单抗也刺激EGFR的内化,消除细胞膜表面的EGFR,从而阻止与配体的反应。除此之外,作为一个IgG1单克隆抗体,西妥昔单抗的ADCC效应(抗体依赖性细胞介导的细胞毒性)还发挥着抗肿瘤的免疫效应:抗体Fab段与肿瘤靶细胞表面的抗原决定簇特异性结合后,同时西妥昔单抗Fc段与NK细胞上的CD16受体结合,可以诱导NK细胞的活化和对肿瘤靶细胞的裂解*伤杀**。
除了西妥昔单抗,也有其它一些抗EGFR单克隆抗体在头颈鳞癌中进行探索,包括帕尼单抗(Panitumumab),扎鲁木单抗(Zalutumumab),尼妥珠单抗(Nimotuzumab)。这些抗体中,帕尼单抗有3期研究去探索在局部晚期和复发转移SCCHN的治疗疗效与安全性,但是很遗憾都是阴性的研究结果,没有为这些疾病阶段的患者带来有效的临床获益。一些研究者认为这是因为帕尼单抗是IgG2单抗,不能诱发ADCC效应或者NK细胞活化后的免疫效应。
但是同样作为IgG1的单克隆抗体,在含铂治疗失败的R/M SCCHN患者使用扎鲁木单抗联合化疗对比单纯化疗治疗的3期研究没有达到主要研究终点(中位总生存),中位无进展生存也仅提高1.5周。3期研究DAHANCA 19中,扎鲁木单抗联合放疗与尼莫拉唑治疗局部晚期SCCHN,提示扎鲁单抗的加入并没有改善局部控制率与生存数据。
尼妥珠单抗也是靶向EGFR的人源化的IgG1单克隆抗体,目前在FDA/EMA均无适应症获批用于SCCHN的治疗(在中国联合放疗仅获批用于EGFR阳性的局晚期鼻咽癌的治疗),也缺乏在复发转移SCCHN中的3期研究探索。另外,在局晚期SCCHN中,尼妥珠单抗联合放化疗对比单纯放化疗的3期研究(NCT01345084)由发起公司撤回;评估CRT±尼妥珠单抗在术后辅助治疗阶段的3期研究还没有结束(NCT00957086)。
02 靶向EGFR的小分子*制剂抑**(TKI)
EGFR-TKI能进入细胞内作用于EGFR的胞内区,干扰ATP结合,抑制酪氨酸激酶活性,阻断激酶的自身磷酸化及底物的磷酸化,以及异常的酪氨酸激酶信号转导。在SCCHN中有探索的EGFR-TKI包括吉非替尼,厄洛替尼,阿法替尼,达可替尼,拉帕替尼。
-
吉非替尼(Gefitinib)
一项2期研究显示,吉非替尼治疗R/M SCCHN,疾病控制率DCR可达53%,但是随后3期临床研究来比较吉非替尼和甲氨喋呤时,并没有得到吉非替尼延长OS,改善ORR的结果。同样,吉非替尼联合多西他赛的3期研究E1302研究也未证实对比多西他赛单药有OS,TTP或ORR的改善。
-
厄洛替尼(Erlotinib)
早期的3期研究探索了厄洛替尼(NCT00412217)作为术后维持治疗或1L治疗R/M SCCHN的研究(NCT00448240),但是因为入组太慢都提前关闭了研究。之后2期研究证实,厄洛替尼相比于放化疗没有改善PFS和ORR。
这些研究提示,单靶点的吉非替尼与厄洛替尼,不论是单药还是联合,对于SCCHN患者都带来了非常有限的临床获益。
-
阿法替尼(Afatinib)
阿法替尼可不可逆的抑制EGFR和HER2。在探索性的2期研究中, 阿法替尼对比西妥昔单抗来治疗含铂治疗失败后的R/M SCCHN,看到了相似的疗效,但是很多患者因为阿法替尼严重的药物相关的AE而停止治疗(23% vs. 5%)包括3级以上的药物相关的皮疹/*疮痤**,腹泻,口腔炎/粘膜炎。3期研究LUX-Head&Neck 1评估了阿法替尼对比甲氨喋呤在含铂治疗失败后的R/M SCCHN,可以看到阿法替尼改善了PFS (2.6 vs 1.7个月; HR = 0.80)以及疾病控制率DCR (49% vs 39%),但没有观察到OS的改善,最常见的3/4级AE包括皮疹/*疮痤**和腹泻。
-
达可替尼(Dacomitinib)
达可替尼也是一种不可逆的泛Erb*制剂抑**,已公开发表的2期研究提示达克替尼一线治疗 R/M SCCHN 患者有部分临床活性,12.7%的患者取得部分缓解。达可替尼治疗最常见的3级以上的药物相关不良反应为腹泻、*疮痤**样皮炎和疲劳,11.6% 的患者因药物相关不良反应而中止研究。
-
拉帕替尼(Lapatinib)
拉帕替尼是一种可逆的EGFR/Erb2双靶点*制剂抑**。一项小样本的2期研究对初治的LA SCCHN患者评估CRT之前使用拉帕替尼,显示相比于安慰剂组,拉帕替尼组患者肿瘤细胞增殖率下降,ORR为17%对比0%。另外一个2期研究中拉帕替尼联合同步CRT时仅看到有略微改善PFS和OS的趋势。一项针对SCCHN高危患者术后拉帕替尼来联合CCRT的3期研究显示,拉帕替尼和安慰剂在无病生存方面没有显著差异,并且拉帕替尼治疗的患者中,48%发生了严重的不良反应。对于R/M SCCHN患者的2期研究中,对于不管是否经EGFR-TKI治疗过的RM SCCHN患者,拉帕替尼治疗都未观察到PR或CR。最新的一个研究评估拉帕替尼联合卡培他滨治疗有过化疗或者放疗史的RM SCCHN,ORR可以达到21%,最常见的3/4级AE为脱水、腹泻、手足综合征(各11%)。
头颈部肿瘤的抗EGFR治疗关键数据总结
参考文献:
1.Bonner JA, Harari PM, Giralt J, et al. N Engl J Med.2006;354(6):567–578.
2.Burtness B, Goldwasser MA, Flood W, et al. J Clin Oncol. 2005;23 (34):8646–8654.
3.Vermorken JB, Mesia R, Rivera F, et al. N Engl J Med. 2008;359(11):1116–1127.
4.Vermorken JB, Stöhlmacher-Williams J, Davidenko I, et al. Lancet Oncol. 2013;14(8):697–710.
5.Stewart JS, Cohen EE, Licitra L, et al. J Clin Oncol. 2009;27 (11):1864–1871.
6.Argiris A, Ghebremichael M, Gilbert J, et al. J Clin Oncol. 2013;31(11):1405–1414.
7.Harrington KJ, Temam S, D’Cruz A, et al. J Clin Oncol. 2014;32(5s):Abstract 6005.
8.Machiels JP, Haddad RI, Fayette J, et al. Lancet Oncol. 2015;16(5):583–594
9.https://science.sciencemag.org/content/367/6484/1307.full
10.https://www.nobelprize.org/prizes/medicine/1986/cohen/lecture/
CHN/ERBMCRC/0320/0092d