▎药明康德内容团队编辑
玩游戏可以治病?FDA批准首款基于游戏的数字疗法
美国FDA宣布,批准第一个基于游戏的数字治疗设备上市,以改善注意力缺陷多动障碍(ADHD)儿童的注意力功能。这种基于游戏的设备名为EndeavorRx,由Akili Interactive公司开发。它只能通过医生处方获得,适用于8-12岁患有注意力不集中或表现出注意力问题的混合型ADHD儿童患者。 EndeavorRx是首个旨在改善ADHD相关症状的数字疗法,也是首个获得FDA批准基于游戏治疗任何疾病的数字医疗手段。对于正在蓬勃发展的数字医疗领域,这是一个重大突破。EndeavorRx是一款能够从网上*载下**的App。它预计将与临床医生指导的疗法、药物治疗,以及教育项目一起,作为整体治疗计划的一部分,改善患者的疾病症状。ADHD是一种开始于儿童期的常见疾病,影响着大约400万6-11岁的儿童。症状包括难以集中注意力、难以控制行为和极高的活动水平。

▲接受EndeavorRX(AKL-T01)治疗患者在TOVA参数上获得改善(图片来源:Lancet Digital Health)FDA审评了包含超过600名儿童的多项研究中获得的数据,这些研究检测了多种与注意力功能改善相关的指标,包括注意力变量测试(TOVA)评估结果、学业成绩评估结果和使用其他评估工具获得的结果。其中一项研究已经在Lancet Digital Health杂志上发表。
靶向小细胞肺癌!FDA加速批准创新疗法
Jazz Pharmaceuticals与合作伙伴PharmaMar联合宣布,美国FDA加速批准Zepzelca(lurbinectedin)上市,用于治疗接受铂类药物化疗时或化疗后疾病进展的转移性小细胞肺癌(SCLC)成人患者。这一加速批准是基于Zepzelca在2期临床试验中的总缓解率(ORR)和缓解持续时间数据。
Zepzelca又名PM1183,是一种烷基化药物,可结合DNA内的鸟嘌呤碱基。这触发了一系列级联反应,可影响DNA结合蛋白(包括一些转录因子)的活性,以及DNA修复通路,从而导致细胞周期的破坏和最终的细胞死亡。去年12月,Jazz Pharmaceuticals公司与PharmaMar达成数额为10亿美元的合作,获得lurbinectedin的开发和推广权益。而中国的绿叶制药集团也在去年与PharmaMar达成合作,获得lurbinectedin在中国的开发及推广权益。
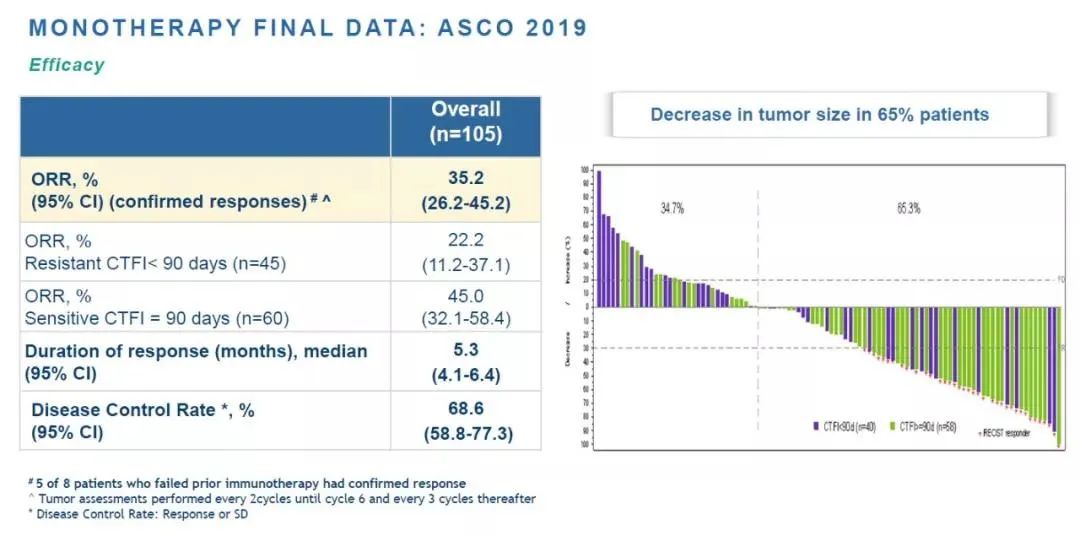
▲Zepzelca的主要疗效数据(图片来源:PharmaMar官网)
Zepzelca的批准是基于一项开放标签、多中心、单组研究的临床数据。该研究在105例经含铂化疗治疗后出现疾病进展的成人SCLC患者中开展。在2020年5月发表在Lancet Oncology杂志上的数据显示,在复发性SCLC患者中,Zepzelca的ORR为35%,研究者评估的中位缓解持续时间为5.3个月。独立审查委员会(IRC)的评估分别为30%和5.1个月。
肿瘤突变负荷首获FDA批准辅助治疗选择!Keytruda第二项“不限癌种”适应症获批
美国FDA官网信息显示,FDA加速批准默沙东公司(MSD)开发的重磅PD-1*制剂抑**Keytruda扩展适应症。用于单药治疗肿瘤突变负荷高(TMB-H)且既往治疗后疾病进展的无法切除或转移性实体瘤患者,无需考虑癌症类型。这些患者的肿瘤组织中TMB≥10个突变/百万碱基,经过既往治疗后出现疾病进展,并且没有令人满意的替代治疗方案。这是肿瘤突变负荷首次获得FDA批准成为指导患者治疗选择的生物标志物。这一批准,有望进一步扩展Keytruda,以及其它PD-1/PD-L1*制剂抑**治疗的患者范围,造福更多癌症患者。此前,Keytruda在2017年获得FDA批准,成为全球首个基于生物标志物的“不限癌种”癌症疗法,治疗微卫星不稳定性高(MSI-H)或错配修复缺陷(dMMR)实体瘤。MSI-H是由于错配修复缺陷,导致DNA的特定区域出现非常多的基因突变。MSI-H的患者对PD-1/PD-L1*制剂抑**的响应比例显著提高,达到约50%。然而,除了结直肠癌和子宫内膜癌以外,其它癌症类型中出现MSI-H的比例并不高,因此使用MSI-H对患者进行筛选,能够影响到的患者人数并不多。以往的研究表明,在TMB-H的患者中,同时出现MSI-H的患者比例在15%~40%之间(如何定义TMB-H对这一数值影响很大),因此如果能够使用TMB-H作为筛选癌症患者的分子生物标志物,那么检查点*制剂抑**就可能造福更多患者。

▲Keytruda药物标签更新部分(图片来源:FDA)这一批准是基于名为KEYNOTE-158的多中心、多队列、非随机、开放标签试验。在这项试验中,不同类型的经治转移性实体瘤患者接受Keytruda单药治疗。TMB-H患者的总缓解率(ORR)达到30.3%,包括4%完全缓解和26.3%部分缓解。而在非TMB-H患者中,ORR为6.7%。在一年后,TMB-H患者中无进展生存率为26.4%,而非TMB-H患者中,无进展生存率为14.1%。而在总生存期(OS)方面,TMB-H患者的中位OS(11.7个月)反而短于非TMB-H患者(13.0个月)。这里值得注意的一点是TMB-H患者携带的肿瘤由于基因突变更多,可能进展更快或更具有侵袭性,从而导致患者预后比非TMB-H患者更为不良。在个体化疗法的时代,医生的目标是通过对患者进行更深入的分析,找到最适合他们的疗法。而基于对肿瘤突变状态的分析筛选最适合免疫检查点*制剂抑**治疗的患者,是提高免疫检查点*制剂抑**疗效的重要一环。在今年的ASCO年会上,默沙东公司公布的临床试验结果显示,Keytruda作为一线疗法,在治疗MSI-H/dMMR结直肠癌患者时,与化疗相比,将患者的无进展生存期提高了一倍(16.5个月比8.2个月)。这意味着基于MSI-H/dMMR这一生物标志物的治疗方法,有望跻身一线疗法。
瞄准“合成致死”!葛兰素史克联手IDEAYA
IDEAYA Biosciences和葛兰素史克公司(GSK)联合宣布,将在“合成致死”领域建立战略合作伙伴关系。合成致死是肿瘤学疗法开发的新兴领域。这一战略合作伙伴关系将包含IDEAYA公司的三个合成致死研发项目,靶向MAT2A、Polθ和Werner解旋酶。预计这些研发项目将在未来三年内达到临床开发阶段。IDEAYA已经解析了MAT2A、Polθ和Werner解旋酶项目的相关晶体结构,让基于结构的药物设计成为可能,其MAT2A和Polθ研发项目已经在相关动物模型中获得了体内概念验证。
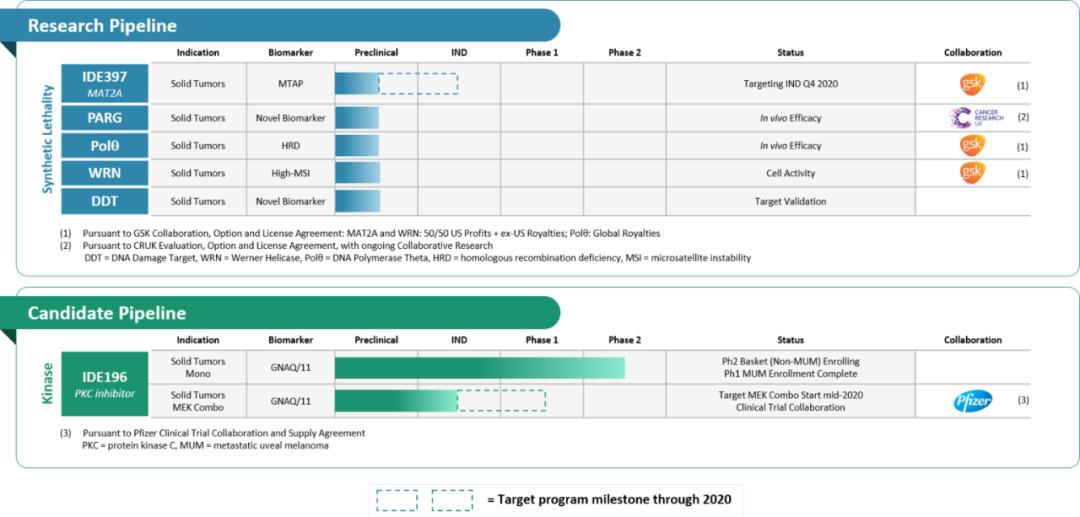
▲IDEAYA研发管线(图片来源:IDEAYA官网)
根据协议,IDEAYA将主导MAT2A项目的早期临床开发。IDEAYA负责在GSK行使选择权之前MAT2A项目的所有费用。此后,IDEAYA将承担20%的全球开发成本。双方还将探索IDEAYA研发项目和GSK研发项目之间的组合。 IDEAYA将获得1亿美元的前期现金付款,以及2000万美元的股权投资。IDEAYA还有权获得潜在的临床前、临床期和销售里程碑付款。
亮剑中轴型脊柱关节炎!诺华重磅IL-17A*制剂抑**斩获第四项FDA适应症
诺华(Novartis)公司宣布,美国FDA已批准该公司的重磅IL-17A*制剂抑**Cosentyx(secukinumab)扩展适应症,治疗活动性非放射学中轴型脊柱关节炎(nr-axSpA)。这是Cosentyx获得FDA批准的第四项适应症。此前,它已经获得FDA批准治疗银屑病、银屑病关节炎和强直性脊柱炎。
Cosentyx是首个可以直接抑制IL-17A的全人源生物制剂。IL-17A是参与银屑病、银屑病关节炎、强直性脊柱炎,以及nr-axSpA的炎症产生及疾病进展的核心致病因子,在发病机制中发挥关键性作用。Cosentyx能特异性结合任何来源的IL-17A,而且不妨碍其他细胞因子的正常工作。因为靶点精准,加上全人源抗体带来的更高安全性,该药在治疗炎症性疾病中体现出快速、持久的疗效及安全性。

Cosentyx的获批基于名为PREVENT的3期临床研究的疗效和安全性结果。该研究纳入了555例活动性nr-axSpA成人患者,这些患者首次接受生物制品疗法,或对抗肿瘤坏死因子-α治疗应答不足或不耐受。试验结果显示,接受Cosentyx治疗的患者在接受治疗52周时,与安慰剂相比,达到ASAS40标准的患者比例获得统计学显著性改善。此外,对患者生活质量和健康状况的问卷评分和其它检测指标也得到改善。
早期三阴性乳腺癌患者福音!罗氏Tecentriq组合疗法达到主要终点
罗氏(Roche)宣布,其PD-L1*制剂抑**Tecentriq(atezolizumab)联合化疗,在治疗早期三阴性乳腺癌(TNBC)患者的3期研究IMpassion031中达到了病理学完全缓解(pCR)的主要终点。Tecentriq联合化疗与安慰剂联合化疗相比,病理学完全缓解(pCR)表现出统计学意义和临床意义的改善,无论患者的PD-L1表达水平如何。罗氏表示,将在即将召开的医学会议上公布具体研究结果,并将与包括美国FDA和欧洲药品管理局(EMA)在内的全球监管机构沟通这次试验的数据。
IMpassion031研究是首个在早期TNBC中展示获益的Tecentriq研究。它是一项随机双盲的多中心3期临床试验,包含333名早期TNBC患者。试验的主要终点是在意向治疗(ITT)人群和PD-L1阳性人群中评估pCR。研究结果显示,与对照组相比,接受Tecentriq组合疗法作为新辅助治疗(手术前)的患者中,无论PD-L1表达如何,在手术时可检测到肿瘤组织的患者较少。新辅助治疗可以让医生快速评估药物是否有效,也可以缩小肿瘤,以便于手术切除。pCR是衡量新辅助治疗效果的常用指标,在早期乳腺癌中可以比传统终点更快评估疗效。

Tecentriq联合nab-紫杉醇目前已在包括美国和整个欧洲在内的70多个国家/地区获得批准,用于治疗肿瘤表达PD-L1(IC≥1%)的无法切除的局部晚期或转移性TNBC的成人患者。在中国,Tecentriq(阿替利珠单抗,商品名:泰圣奇)于今年2月份获批,联合化疗用于一线治疗广泛期的小细胞肺癌。
重要突破!口服微生物组疗法获得首个关键性临床试验积极结果
Finch Therapeutics公司宣布,该公司开发的研究性口服微生物组药物CP101,在预防复发性艰难梭菌感染(CDI)的多中心、随机双盲、含安慰剂对照的2期临床试验中获得积极顶线结果。新闻稿指出,这是口服微生物组疗法首次在关键性临床试验中获得积极结果,是微生物组疗法开发领域的重要突破。

研究表明,肠道微生物组功能障碍与一系列严重疾病的发病机制相关。CP101以口服肠溶胶囊形式递送完整的微生物群落。旨在通过恢复定植抗性,或健康的微生物组,来防止潜在病原体定植的能力,从而预防复发性CDI。它已经获得美国FDA授予的快速通道资格和突破性疗法认定,用于预防复发性CDI。 在名为PRISM3的临床试验中,CP101达到了主要疗效终点,接受CP101单次给药的复发性CDI患者中74.5%在第8周达到了持续的临床治愈,与对照组患者的61.5%相比,改善具有统计学意义(p<0.05)。CP101在研究中耐受良好,无治疗相关严重不良事件。