
罕见病之痛
今年正值国际罕见病日设立10周年。我国公布的《第一批罕见病目录》,为罕见病患者的健康保障打开了一扇希望之门。未来,要使罕见病患者免于不幸,仍需社会各界的协同努力。
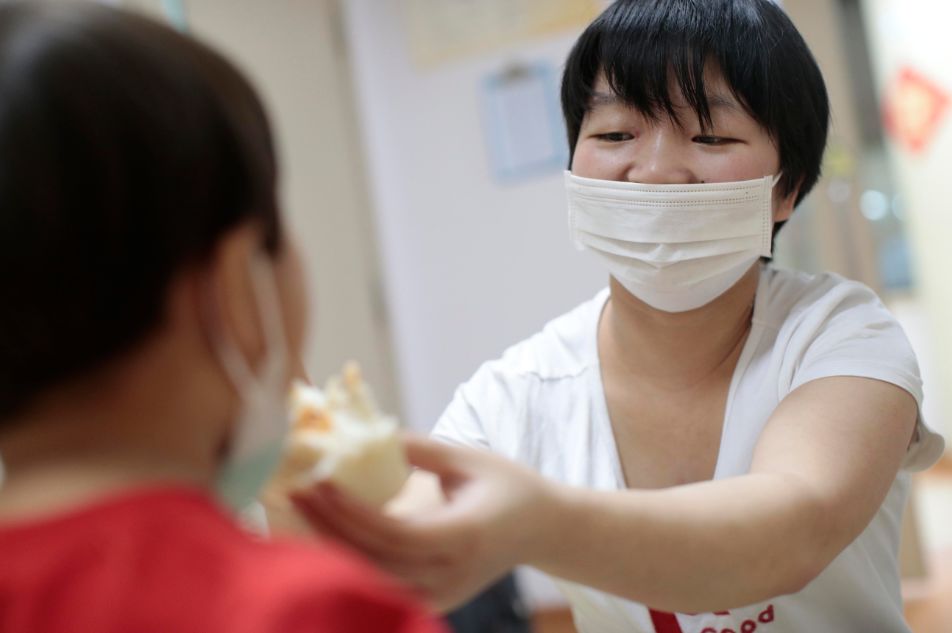
2017年9月,上海。一位罕见病男童骨髓移植
两度失败,3岁的姐姐坚持要捐骨髓救弟弟。
图为妈妈在给姐姐喂饭。
“在蝴蝶结病治疗上,
是病人推着我们往前走”
“我做医生这么多年,唯独在蝴蝶结病的治疗上,是病人推着我们往前走。”说起治疗罕见病的体会,解放军总医院儿内科主任、中国人民解放军儿童医学中心主任邹丽萍感慨道。
2009年,邹丽萍第一次接诊蝴蝶结病患儿。此前,蝴蝶结病只是她医学专业知识储备里一个抽象的存在。
邹丽萍仍记得辽宁省大连市患儿小翼(化名)刚到解放军总医院住院时的情形,“孩子当时才1岁,抽得很厉害,最多时一天抽200多次。小孩的肝功能出现异常,对药物也有抵抗”。
患儿父亲陈先生说,孩子从出生起就有癫痫发作,大连当地医院怀疑是蝴蝶结病,到解放军总医院后才得以确诊。
蝴蝶结病的专业名称是结节性硬化症。在5月11日由国家卫生健康委员会等5部门联合发布的《第一批罕见病目录》里,结节性硬化症名列其中。
邹丽萍说,蝴蝶结病是一种常染色体显性遗传病,是由于TSC1或TSC2基因突变引发雷帕霉素靶蛋白(mTOR)信号通路异常活化,最终导致细胞异常分化和增生并累及多个器官的综合征。患儿往往在胚胎时期就会出现心脏肿瘤,出生以后皮肤会有白斑,并有癫痫发作,再大一点会有脑部、肝脏、肾脏的肿瘤等,还可能出现自闭症。其中,癫痫对患儿的生命和生活都有严重影响,可能造成智力受损,这主要是由于大脑皮层某些发育不良,形成皮层结节(结节性硬化症命名由来),影响了大脑正常的“信号传递”,从而导致出现抽搐。由于目前尚缺乏有效的治疗手段,一些患儿因没有得到及时治疗,而出现严重后果甚至死亡。
“我从来不会说‘这病治不了’‘这病没法治’,而是想着怎么样能帮助患者。”对于小翼,邹丽萍努力探寻治疗的办法。
因为蝴蝶结病病因主要出在mTOR信号通路上,而与这个点对应的药雷帕霉素可能对治疗有帮助。“正好我们医院有这种药,那为什么不试试呢?”邹丽萍跟患儿家长做了详细的沟通,同时把治疗的风险和可能的不良后果和盘托出。
邹丽萍的诚恳打动了患儿的家长,他们当即表态:“我们愿意试试。”
邹丽萍先向医院的伦理委员会申请,得到批准并在医疗处备案,然后和患儿家长签订了知情同意,开始尝试这种试验性疗法。
令人欣慰的是,用雷帕霉素治疗蝴蝶结病伴发的癫痫取得了不错的效果。小翼的癫痫症状基本得到控制——抽得次数明显减少,脑电图显示好转。“我们一直用这个药进行治疗,定期复查,到现在小孩快10岁了,基本不抽了,看上去跟正常小孩没什么两样。”说起孩子的近况,陈先生对邹丽萍满怀感激。
邹丽萍把治疗的成功归结为“西医的诊断,中医的思维”,即把患者当作一个整体的动态的人来看待,而不是只看到某些症状进行治疗。她说:“癫痫、皮肤白斑、肿瘤等都是蝴蝶结病的症状,是这个病在不同生命周期的表现,而这些症状在一些常见病上也可能有。如果我们只是看到某种症状,只会对症治疗,就永远找不到病因,也就不可能真正治疗这种罕见病。”
“进行试验性疗法,
我更关心病人能否获益”
孩子得救后,陈先生把小翼的案例发在了病友群里,陆续有更多的蝴蝶结病患儿家长慕名来找邹丽萍治疗。这些不断增多的案例,为她研究蝴蝶结病积累了丰富和宝贵的资料。
2014年,邹丽萍在《中华儿科医学杂志》上发表了《雷帕霉素治疗结节性硬化症合并癫痫的临床效果和安全新观察》一文。文章通过对52名患儿的对照研究,得出结论:雷帕霉素治疗结节性硬化症合并癫痫的疗效确切,不良反应少。
雷帕霉素是器官移植手术后使用的抗排异药物,蝴蝶结病并不在该药的适应证之列,邹丽萍的探索拓展了雷帕霉素的临床用途。
也是在这一年,罕见病全球学术科研会议首次在北京召开。会上,邹丽萍分享了在治疗蝴蝶结病用药和诊疗上的一些新思路和方法。她从一直致力于蝴蝶结病科研的北京协和医院张宏冰教授那里得知,虽然国际上已有关于雷帕霉素治疗蝴蝶结病合并癫痫的文献报道,但最早运用在临床上,她是第一人。很多人劝她发英文文章,但邹丽萍为了告诉国内同行蝴蝶结病有药可治,连续发表3篇中文文章,还在不同的会议上推广宣传。
治疗蝴蝶结病的独到经验让邹丽萍在业界以勇于探索而闻名。然而,她的老药新用这一试验性疗法也招致业界一些非议,有人甚至给她扣上“非法行医”的帽子。面对罕见病无药可治的情形,医生尝试老药新用,能否过法律关、伦理关,不只是邹丽萍一个人面临的窘境。
对于医生面临的风险,清华大学附属长庚医院医患办覃涛认为,如果按照我国《处方管理办法》第14条规定和《医疗机构药事管理规定》第18条的规定,超说明书用药有违规之嫌,但我国《执业医师法》也规定“医师进行试验性临床医疗,应当经医院批准并征得患者本人或者其家属同意”。由此可见,超说明书用药这种试验性治疗并非没有法律依据,“对于某些罕见病无药可医的情况,这种老药新用的方式具有很大的临床意义”。
中国医学科学院生命伦理学研究中心执行主任翟晓梅教授则表示:“医生出于患者最佳利益的考虑,基于对药物作用有一定的了解和把握的基础上,尝试老药新用在伦理上是可以得到辩护的。”
说起探索的动力,邹丽萍说:“我是一个临床医生,我进行试验性疗法,除了做科研,更关心的是病人能否获益。”不只是蝴蝶结病,目前85%的罕见病是基因病,缺乏有效的治疗方法,很多医生在接诊罕见病患者时都束手无策。
邹丽萍的同事、解放军总医院儿内科副主任医师孟岩,在治疗罕见病戈谢病方面也颇有经验。看到患者因用不起昂贵的进口药,孟岩会选用一些便宜且有效的国内药物进行治疗,这同样超出了所用药的说明书适应证范围。她担心的是,“万一出现问题,患儿家长闹起来,怎么办”。孟岩呼吁:“我国能否像美国一样,出台与《孤儿药法案》类似的有关罕见病诊治的法律,至少是政府颁发相关的文件,鼓励医生创新性治疗,而不是让医生孤身承担治疗风险。”
“不但要治病,
还要帮助患者解决经济困难”
今年5月公布的国家版第一批罕见病目录纳入了121种罕见病。消息出来后,罕见病患者群体中一片欢欣鼓舞,但也有一些人对白塞病、神经纤维瘤病等没有入选目录表示不解。
作为参与该名录拟定的专家组成员,邹丽萍解释,第一批罕见病筛选,一是参照国际标准,再有就是根据国情,选择那些目前有些方法能治疗的罕见病,实现延缓病情发展或者根治。
有成熟的方法、合适的药物,为什么不能用于治疗更多的患者,让更多的患者受益呢?基于这种想法,邹丽萍除了在解放军总医院治疗罕见病,还毫不保留地把经验传授出去。比如,她在山东省临沂市人民医院小儿神经科建立了蝴蝶结病专科诊疗点,每年她都会去讲课和带教基层医生。
临沂市人民医院小儿神经科主任、儿童癫痫中心主任李玉芬说:“现在,我们小儿神经科的医生,在接诊癫痫患儿时,都会留心查看患儿身上的皮肤有没有白斑。如果查出是蝴蝶结病,就能做到早干预。在用雷帕霉素进行治疗时,我们按照邹主任给提供的模板,签订知情同意,进行规范治疗。”
横亘在罕见病患者面前的另一大难关是高昂的费用问题。邹丽萍在用雷帕霉素给患者进行试验性治疗时,通常会给患者申请减免费用,但这只是杯水车薪。如果能把雷帕霉素纳入蝴蝶结病治疗的医保中,就可解决患者有药用不起的问题。
“从2009年到现在,我们已经有1400多例的临床数据证明了雷帕霉素治疗的有效性和安全性,这足以支持把蝴蝶结病的适应证加到雷帕霉素的说明书中。雷帕霉素一盒只要1000多元,而用于治疗蝴蝶结病的另一种有效的进口药依维莫司,一盒要6000多元。我们为什么不支持自己的医药呢?”邹丽萍很是着急。
“一些经济发达的地区可以考虑把一些罕见病用药纳入医保。对于超说明书用药的老药,如果已纳入医保,则可以通过形成专家共识,扩大适应证,使之成为罕见病的医保用药。另外,对于一些不是很贵的罕见病用药,也可以纳入医保。”翟晓梅教授提出,“实际上,国外针对罕见病研发的药,往往价格非常昂贵,如果都纳入医保,对于普通老百姓来说是不公平的,毕竟国家用于医保的财力是有限的,肯定是要先保障绝大多数人的基本医疗权利。但是,如果国家财政完全不管罕见病患者,对他们也有失公允。我认为,解决罕见病患者的用药难问题,需要依靠国家、社会和个人共同努力,可以通过国家的大病统筹给解决一部分费用,社会支持如慈善也出一部分力,家庭也要承担一部分责任。同时可引入商业医疗保险作为补充。”
为了帮助更多患者,邹丽萍在由解放军总医院发起的北京市儿科专科医联体的推动下带领团队入驻京都儿童医院,成立神经与罕见病科。她把在京都儿童医院的劳动所得,全部捐给中华少年儿童慈善救助基金会爱心家园救助中心,并借助社会爱心人士的帮助,首次助资500万元成立以她名字命名的罕见病公益基金。“这样,我们不但能够治疗罕见病,还可以用基金的钱帮助罕见病患者,支持有创新性的研究。这是我今后10年的目标。”邹丽萍说。
罕见病之痛
小郭:
有药我们却用不起
2011年,郭朋贺大学毕业,开始了充实而快乐的北漂生活。幸福的小日子刚刚起步,却在2014年戛然而止。这一年,郭朋贺被诊断为庞贝氏症。
庞贝氏症,又名肝醣储积症第二型,是一种十分罕见的先天性疾病,中国的发病率约为五万分之一。该病患者体内缺少酸性α-葡萄糖苷酶,使体内肝糖原无法被分解利用。肝糖原是人体内贮藏能量的“罐头”,运动时可为肌肉提供能量,庞贝氏症患者身体里缺少的酶是打开“罐头”的工具。缺少了这种酶,就使得肝糖原无法被利用,患者会出现肌肉萎缩等一系列症状。
病情一天天发展,郭朋贺消瘦、无力的症状不断加重。身高1.6米的她,如今只剩下30千克体重,对抗自身重力成为最大的难题,出门走一走都是一件值得在病友间炫耀的事。
郭朋贺手机里有一个庞贝氏症患者微信群,里面有127位病友,郭朋贺是他们中的“运动达人”。她说,希望其他病友看到她,也能鼓起勇气,出去走走。但是,当她睡觉时,由于呼吸肌群不能抵抗重力、支撑起胸腔,就不得不借助呼吸机维持呼吸。她说,呼吸机是庞贝氏症病友床头的标配。
郭朋贺最害怕冬天。2017年冬天,群里有6位病友离开人世,都是因为感冒。感冒后呼吸道产生的分泌物,正常人能通过咳嗽排出,但庞贝氏症患者无力咳痰,很容易窒息而死。
美而赞是当前唯一可以阻止庞贝氏症病情发展的药物,但需要患者终生使用。“只要足量用药,庞贝氏症患者和正常人没什么两样。”郭朋贺说。
但有药却用不起。“以我现在30千克的体重算,足量用药的话每年费用要200多万元。用药后体重增加,药量也要增加,费用会达到每年300万元。”郭朋贺说,她认识的100多位病友中,只有9个患者在用药,其中仅有的两个成年人,也都不是足量用药。
发病机理和治疗费用都与庞贝氏症类似的还有戈谢病。在社会关注下,多地都出台了戈谢病患者救助政策,患者大多都在积极治疗。但目前,治疗庞贝氏症的美而赞尚未进入医保。一些地方性保障政策把庞贝氏症用药纳入保障,但条件十分苛刻。比如每年有20万的报销上限,以及患者家庭必须是民政部门认定的低保户等。而一个成年人每年的药品总费用在200万元以上,20万元的报销上限远远不能满足用药需求。
不用药成为绝大多数庞贝氏症患者的选择。
近期,国家卫生健康委员会公布了第一批罕见病目录,庞贝氏症在列其中,这让郭朋贺看到了希望。“期待国家能依据目录出台相应的保障政策。我们等得太久了。”郭朋贺说。
小马爸爸:
花了30万,治疗方向都不对
2012年春天,东北汉子马志强2岁的儿子小马身上长了疹子。当地医院看过之后认为没什么大问题。可没多久,小马头部又出现了包块,市区医院医生诊断为气泡瘤。
小马陆续出现了眼球突出、骨损伤、中枢性尿崩症等症状,淋巴结和肺部也发生了病变。马志强带着小马从东北到北京,辗转十几家医院的不同科室,收到两次病危通知书。虽然孩子顽强地挺过来了,但病因始终是个谜。
马志强当时并不知道,儿子身上的各种症状其实是一种病的并发症。朗格罕斯组织细胞增生症(LCH),一种发展迅速的肿瘤性疾病。患者骨髓来源的朗格汉斯细胞增生,进而累及多个器官。这种病的发病率为二十万分之一至二百万分之一。
“第一次听到这个名字是在哈尔滨一家三甲医院。”马志强回忆说,“当时医生翻着厚厚的医书,说这些症状疑似朗格罕斯组织细胞增生症,但也不能确定。”
马志强开始上网搜索这种从没听说过的怪病。最终,他找到了一个朗格罕斯组织细胞增生症患者的QQ群。热心的群友给他指了一条路:去首都儿研所,找血液病科师晓东主任。
揣着东借西凑的几万元钱,马志强抱着孩子再一次来到北京。经过基因筛查,最终确定,小马患的就是朗格罕斯组织细胞增生症。师晓东为小马制订了一套完整的治疗方案。经历了45次化疗之后,小马终于痊愈了,现在仅需要对偶尔复发的并发症进行对症治疗。
“能早点确诊就好了。”马志强说,儿子从发病到确诊,前后用了1年多时间,跑了十几家医院,耽误了最佳治疗时间。等到确诊时,一些并发症已经造成了器官损害。
“确诊以后真正治病花的钱其实还不到5万元。”马志强说,“前面花了30多万元,治疗方向都不对。”
师晓东说,随着基因疗法、骨髓移植技术、精准靶向治疗等医学科技的进步,像LCH、黏多糖贮积症、WAS综合征等罕见病,目前都已有成熟的治疗方案。但由于这些疾病十分罕见,能正确诊断的医生很少。
师晓东说,首都儿研所正在着手筹备面向全国医务人员的培训活动,重点普及罕见病的鉴别和诊断,让更多患者可以尽早得到诊断。
让人性光芒照耀更多患者
罕见病是一个医学难题。在人类攻克疾病的漫长历史中,总会遇到一些疑难怪病,罕见病则是其中更为突兀的“魔怪”。就目前医学发展水平而言,很多罕见病因罕见、难以识别等因素,导致误诊、漏诊时有发生。由于80%的罕见病是基因遗传病,即使诊断明确了,往往也一时难以找到有效的治疗手段,导致罕见病患者只能在痛苦中“熬日子”。这是人们不忍看到的结局,也让很多医生产生了深深的无力感。
如何攻克罕见病?无疑是摆在医学面前的一道难题。我们必须承认医学是有局限性的,但同时,医学也是一门不断发展的科学,是一项人道主义的事业。为了解决罕见病患者“看病难”问题,我们需要邹丽萍这样有担当有责任感的医生,不断为了患者的利益,勇于探索创新,努力开辟可能的救治通道。今年以来,上海、北京等地多家罕见病诊治中心的成立,也让我们看到了医疗界在不断发力。
罕见病还容易衍生出不容忽视的社会问题。对罕见病患者来说,“罕见”二字意味他们的求医之路更加艰难、曲折。一些罕见病用药不但稀少,而且价格昂贵,使得罕见病患者所遭遇的“看病贵”问题尤为突出。为了活命,在国内求药不得的情况下,有些人到境外买药、代购“救命药”;更有一些非法之徒从中嗅到商机,代销违禁药品,从中牟利。
有些罕见病患者在孤苦中建立了自己的内援圈子——通过病友群自然而然地聚合到一起,彼此帮助,互相温暖。但毕竟这种抱团取暖的力量有限,也非长久之计。罕见病给他们带来的巨大风险非个人和家庭所能承受,需要举国家之力、集社会之力避免深陷不幸的泥潭。诚然,受经济发展和社会保障水平等条件的限制,罕见病用药完全进医保尚存在诸多困难。但本着医疗资源的分配公正性原则,对于已经被纳入国家版罕见病目录的罕见病用药,可以有选择地纳入医保。我们也呼吁社会爱心和公益组织加入到帮助罕见病患者的队伍中,让他们感受到人性的温暖和善良。
面对疾病,我们有时无奈,但不能少了温暖。希望国家罕见病目录的发布,能推动医疗技术、治疗药物、费用保障等的进步和完善,让人性的光芒照耀更多罕见病患者。
文/健康报记者 李阳和 姜天一
图/视觉中国
编辑/管仲瑶
原创声明:以上为《健康报》原创作品,如若转载须获得本报授权。
点击下方爱心,您的赞是对我们最大的支持!
↓↓↓