在您开始阅读文章之前,诚恳地请求您点击一下“关注”,您不仅可以方便地参与讨论和分享,还能获得更多的互动体验。感谢您对我们的支持。
文|铭文记事
编辑|铭文记事
引言
催化是化学领域中一个极为重要的研究方向,其在现代化工、材料科学、医药工业以及环境保护等领域中都有着广泛的应用。 传统的催化研究是基于实验的,然而实验条件的限制和复杂性,使得开发新型催化剂和提高催化效率成为一项挑战。 随着计算机科学技术的迅速发展,电子结构计算方法的应用为改善催化研究提供了新思路。
本文将着重探讨电子结构计算方法在催化研究中的应用,包括其在催化反应机理的研究中的作用,以及对催化剂设计和合成的影响 。同时,本文还将介绍未来电子结构计算方法在催化领域的发展趋势,以及在催化研究中存在的局限性和挑战。

电子结构计算方法的基本原理
作为计算化学领域中的一种重要研究手段和方法,电子结构计算方法以其基于量子力学原理的理论模型和计算技术,为我们深入了解分子和材料的微观结构、性质与功能提供了有力的支持。
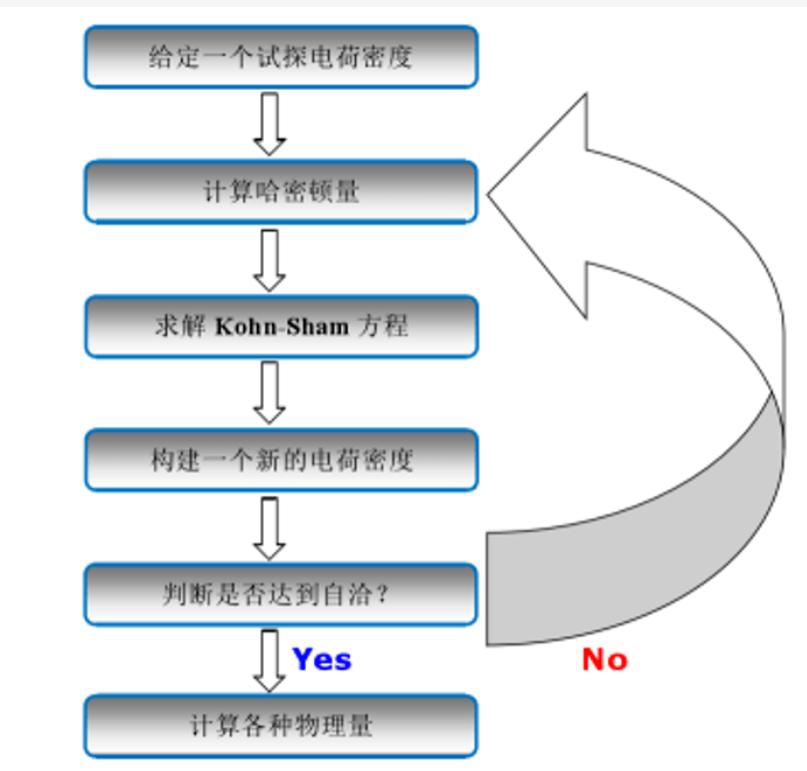
电子结构计算方法基于薛定谔方程,采用量子化学原理来描述单个分子或团簇的电子构型和能带结构,并通过计算化学模型和算法,预测其化学性质和响应行为。其基础理论包括自洽场方法、密度泛函理论和多电子激发理论等。其中,自洽场方法是指通过计算电子的Pauli排斥能和相互吸引能来确定原子轨道以及分子的电荷分布情况,以此计算体系的总能量和其他物理性质。自洽场方法通过SCF(self-consistent field)计算,求解出轨道能量、轨道电子数和Fock算符,从而得到一个由分子轨道组成的HF(Hartree-Fock)波函数,该波函数可以反映分子的基态能量和电子结构。
密度泛函理论(DFT)揭示了电子运动状态与电荷密度之间的关系,是电子结构计算方法中最常用的方法之一。它的基本思想是,将分子的总能量表示为电子密度的某一个函数关系,并通过最小化相应的泛函来表征分子的总能量和其他物理性质。在DFT的计算过程中,需要通过构建有效的交换和相关泛函来描述电子之间的相互作用,以及各自占据的状态和能量等信息。
多电子激发理论涉及到复杂的分子体系,其基本思想是,通过对原子轨道进行线性组合,来构建含有多个电子的激发态波函数,并通过计算相关的哈密顿算符,得到分子体系的基态和激发态能量以及电子的相关行为。
电子结构计算方法在催化领域的应用早已成为主流研究方向。其主要作用包括深入研究有机和无机分子之间的相互作用,探索催化反应的机理和动力学过程,预测和设计新型催化剂或者催化材料,以及优化已有催化剂的性能。在分子和材料合成、功能材料和催化剂材料等领域中,电子结构计算方法不断发挥出其重要的作用。
在催化反应机理研究方面,电子结构计算方法通过计算并描述反应物、中间体和产物之间的电子转移和相关行为,可以揭示反应机理的细节,包括反应物之间的键合,中间物的形成,以及产物的生成过程等。通过电子结构计算,可以预测分子之间的吸附能和吸附结构,合理设计催化剂的反应中心以及调控反应的速率和选择性。
在催化剂设计和优化方面,电子结构计算方法可以帮助我们预测催化剂的晶体结构和化学组成,并揭示其催化活性和稳定性的机理,更好地设计和合成新的催化剂或者改善已有的催化剂的性能。同时电子结构计算方法也可以帮助我们分析和预测材料的光学、电学、磁学和力学性质,为新材料的合成和设计提供支持。
然而,电子结构计算方法的应用还存在一些挑战和局限性。例如,尽管DFT可以实现第一性原理计算,但对于大分子和超分子系统的计算则需要消耗巨大的计算资源和时间,计算结果也易受到初始参数的影响。在处理强关联电子系统和金属表面催化剂中,常规的电子结构计算方法也受到一定的限制。
因此,为了更好地促进电子结构计算方法在催化领域的应用,需要不断加强相关的算法研究和计算资源的开发和优化,同时也需要进一步加深对电子结构计算方法的理论基础的理解和掌握。总的来说,电子结构计算方法在催化领域的应用前景广阔,其在预测和设计新型催化剂、深入研究催化机理等方面具有重要的科学价值和应用价值。
电子结构计算方法在催化反应机理研究中的作用
催化反应机理研究是化学领域中的一个重要方向,它关注分子间相互作用、能量转换和反应速率等核心问题,是探索催化反应内在机理和优化催化剂设计的关键。电子结构计算方法作为一种理论模型和分析工具,具有高精度、高效率和灵活性等优点,为探究催化反应机理提供了可靠的理论基础和计算手段。本文将从以下几个方面来介绍电子结构计算方法在催化反应机理研究中的作用。
1.确定反应机理和确定反应途径
电子结构计算方法能够模拟化学反应过程中电子的态密度和轨道分布情况,同时考虑分子、催化剂等相互作用,可以在原子和电子层面上描述化学反应的机理和途径。通过计算催化剂表面活性位点、反应物和中间体分子的结构构型、能量势能面图等信息,可以预测反应的活化能、化学物种形成和分解的能量变化情况,揭示反应的动力学性质和化学特征,并为寻找催化反应最佳条件和催化剂设计合成提供理论依据。
2.揭示催化剂结构和表面特征
催化剂的结构和表面结构是影响反应机理和性能的主要因素之一,因此了解其表面结构成为探究催化剂反应机理的关键之一。电子结构计算方法能够探究催化剂表面的电子结构、散射横截面、局域化反键轨道等表征指标,预测比表面积、孔隙结构和活性位点等表面相关参数,解释催化剂的反应机理和催化活性,为设计和合成高性能催化剂提供理论指导。
3.分子模拟与反应动力学研究
电子结构计算方法可以利用分子动力学仿真和其他分子模拟技术,扩展化学反应模型的尺寸和范围,同时解决复杂的相互作用和弛豫过程。通过计算分子间距离、偏极性结构、构型和反应速率等参数,可以揭示分子之间的相互作用和重新排列规律,从而进一步推断催化反应体系的结构和动力学过程,为优化反应条件和催化剂设计和合成提供指导。
4.探讨电子结构计算方法的局限性和挑战
在应用电子结构计算方法研究催化反应机理的过程中,仍然存在一些挑战和局限,如计算模型假设、计算精度和计算效率等方面的不足。例如大规模电子结构计算的困难、时间尺度和空间尺度上的限制、计算误差的存在等问题不可避免。因此,为了克服这些局限性和挑战,需要更加深入地研究电子结构计算方法和发展新的计算技术和算法,以期在催化反应机理研究中获得更加深入、全面和准确的理论预测和分子设计指导。
电子结构计算方法对催化剂设计和合成的影响
电子结构计算方法作为一种基于量子化学理论的计算技术,具有高精度、高效率和安全性等优势,被广泛应用于催化剂设计和合成的研究中,能够为化学工业的可持续发展提供有力支撑。本文将从以下几个方面来介绍电子结构计算方法在催化剂设计和合成中的影响。
1.筛选催化剂候选材料和合成路径的优化
催化剂的研究需要筛选大量的候选材料,并优化设计催化剂合成方法。计算化学中,电子结构计算方法可用于模拟不同催化剂的晶体结构、表面活性位点、电子结构与原子间相互作用等与催化反应性能相关的因素,以预测催化剂性能的关键参数和物理指标。这些指标可以帮助化学工程师发现新的催化剂候选材料,并确定最优的催化剂设计方案和合成路径,以提高催化剂的活性、选择性和稳定性。

2.分析催化反应过程的机理和动力学
电子结构计算方法可模拟化学反应过程中的电子结构和能量势能面,为研究催化反应的机理和动力学提供基础的计算模型。通过计算催化剂表面活性位点、反应物和中间体分子的结构构型、能量势能面、反应活性、反应速率和反应途径等指标,可以揭示催化反应的本质和规律,并为催化剂的设计和合成提供指导性意见。
3.优化催化剂性能和稳定性
催化剂的性能和稳定性也是研究的关键问题,使用电子结构计算方法可以预测催化剂的化学稳定性和热力学性质,为催化剂稳定性设计提供基础理论依据。同时,还可以模拟催化剂表面激活并稳定反应物,控制催化剂表面的形态和形貌,提高催化剂的活性和选择性,以及延长催化剂的使用寿命,从而推动催化剂研发和工业应用进程。
4.探索新型催化剂材料
配位量子化学、电子传递理论和高通量计算方法结合使用,可以探索新型催化剂材料,优化催化剂反应活性、产率和选择性等重要综合指标。计算化学的人工智能算法、数据挖掘和模型优化技术,还能帮助化学工程师更快、更智能地发现和预测新型催化剂材料,促进催化剂技术的不断进步和发展。
综上所述,电子结构计算方法在催化剂设计和合成中的作用不可忽视,其高效性和准确性对于推动化学工业的可持续发展的重要性不容置疑。未来,随着计算技术和计算化学理论的不断进步,电子结构计算方法在催化领域的应用前景将更加广阔。
未来电子结构计算方法在催化领域的发展趋势
未来电子结构计算技术的发展趋势在于高精度、高效率、高通量和高自适应性四个方向。随着计算系统性能的提高,计算和模拟的领域将不断拓展。
针对催化研究的需求,在电子结构计算基础上发展的新技术也逐渐得到应用。其中,包括多尺度模拟技术、机器学习算法以及GPU加速智能算法等。这些新技术不仅能够处理更大的分子和更加复杂的催化体系,而且还能够准确地预测反应的活化能、动力学参数和通量,从而进一步完善和拓展催化反应模型。
电子结构计算方法在催化研究中存在的局限性和挑战
电子结构计算方法在催化研究中也存在着许多挑战和限制。例如,电子结构计算方法通常依赖于大量计算程序和复杂的算法,需要强大的计算机支撑。另外,由于涉及到庞大的体系,计算效率通常比较低。同时,电子结构计算方法对不同计算参数的敏感性较高,计算结果的可重复性需要较高的控制。
不过,上述问题也有一些解决方案。例如,可以通过集群技术和GPU加速进一步提高计算效率,使用并行计算和改进的算法降低噪声和误差,提高计算结果的稳定性和准确性等。这些技术的出现显著提高了电子结构计算方法在催化研究中的实用性和可靠性。
作者观点
在现代催化研究中,电子结构计算方法已经成为了一种重要的方法。通过计算反应中间体、势垒、能量面和吸附特异性等,可以揭示催化反应的本质,探索催化机制,优化催化剂设计和合成。未来,电子结构计算方法技术的发展将会进一步推动催化研究的创新,提高催化材料性能和效率,为化学领域的发展做出更大贡献。
参考文献
1. Liu, J., & Li, Y. (2017). Perspective: Opportunities and challenges in electronic structure methods for heterogeneous catalysis. The Journal of chemical physics, 146(20), 200901.
2. Wang, R., Lin, Z., & Li, Y. (2018). Recent advances in computational catalysis: methods and applications. Chemical Society Reviews, 47(23), 8527-8544.
3. Guo, J., & Sun, S. (2016). Computational studies of catalytic reactions: insight into reactions and catalysts. Chemical reviews, 116(12), 7140-7189.
4. Wang, X., Li, Y., & Liu, J. (2019). State-of-the-art computational methods for predicting the structure and properties of catalytic materials. Chemical Society Reviews, 48(2), 525-539.
5. Winkler, B., Hong, W., & Baiker, A. (2016). DFT in heterogeneous catalysis: Theory and experiment. Chemical Society Reviews, 45(15), 4191-4204.