
全文简介
*土稀**过渡金属氧化物(RE-TMO)是OER的前沿研究领域,但是有关其电催化机理和活性位点的知识非常有限。本研究通过有效的等离子体(P)辅助策略成功地设计和合成了原子分散的CoO表面Ce单原子催化剂(P-Ce SAs@CoO),以探讨RE-TMO催化系统中OER性能的起源。P-Ce SAs@CoO表现出良好的性能,在10 mA cm-2时仅需261 mV的过电位,并具有稳定的电化学稳定性,比单独的CoO优越。X射线吸收光谱和原位电化学拉曼光谱揭示了Ce诱导的电子重新分配抑制了Co-O-Ce单元位点中Co-O键的断裂。理论分析表明,梯度轨道耦合通过优化Co-3d-eg占据数,增强了Ce(4f)-O(2p)-Co(3d)单元活性位点的Co-O共价性,从而平衡了中间体的吸附强度,并进一步达到了理论OER极限,与实验观察结果非常一致。我们相信,建立这种Ce-CoO模型可以为理解高性能RE-TMO催化剂的机理和结构设计奠定基础。
结果与讨论
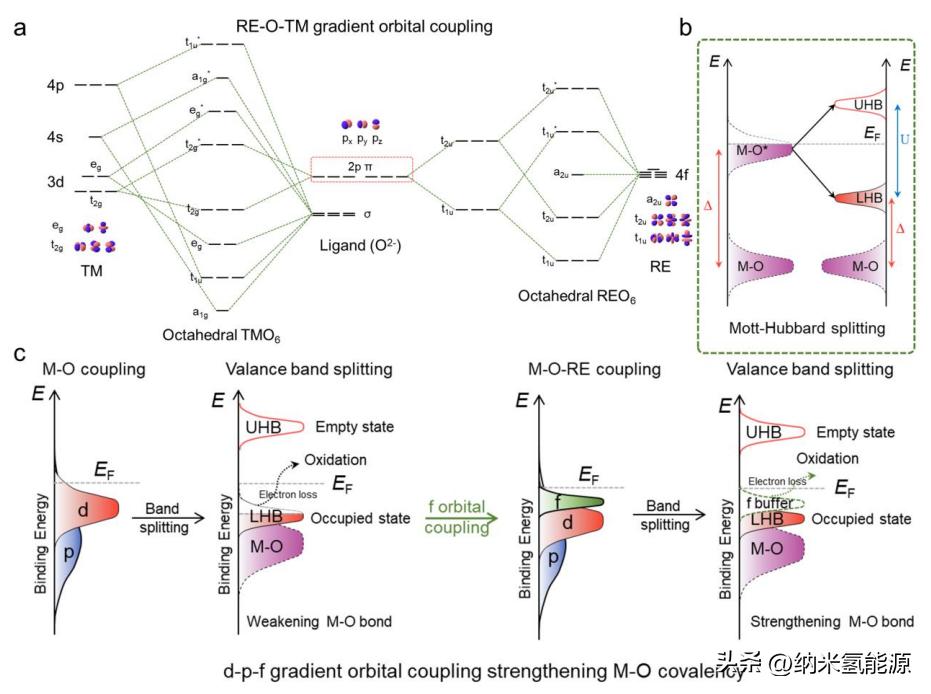
图1 提出了TM-O-RE单元位梯度轨道耦合合理性的理论机制。(a)由[TMO6]和[REO6]得到的具有Oh对称的定性分子轨道图;(b)基于莫特-哈伯德理论的波段结构分裂;(c) Mott-Hubbard分裂中d-p-f梯度轨道与f带电子缓冲耦合的构建。
具有独特[TMO6]和[REO6]边缘的TMOs和REOs共享相同的Oh对称性(图S1)。由于氧原子的σ和π电子对TM(Oh)-O-RE(Oh)的贡献,可以形成TM-O-RE的梯度轨道耦合(图1a),为电催化适应性提供更灵活的电子相互作用(例如,优化eg占据和M-O共价性)。在TMOs中,M-O共价性可以形成M-O键和(M-O)*反键带,M-O键带内的电子灵活占据解释了电子导电性。然而,一些TMOs(例如CoO和NiO)在Néel温度以下呈绝缘体性质,这与带结构理论的预测相矛盾。针对这个问题,莫特-哈伯德效应通过考虑强的原位d-d库仑交换相互作用U,将(M-O)*分裂为填充的低哈伯德带(LHB)和空的上哈伯德带(UHB),从而进行理论校正(图1b)。M-O带和(M-O)*带之间的带隙被称为电荷转移能量(Δ),取决于M和O之间的电负性和马德隆势。需要注意的是,U与d金属种类的轨道体积和价态成反比关系,而U和Δ之间的差异可能会导致靠近费米面处3d电子的贡献差异,从而显示出调节M-O共价性的潜力。通过莫特-哈伯德分裂,如果电荷转移能量可以抵消原位库仑和交换相互作用能(U<<Δ),则充电过程(如OER)将从填充最低带的LHB到M-O(过度氧化)提取电子,这将牺牲M-O共价性和高活性的On-物种(图1c,左侧)。通过构建TM(3d)-O(2p)-RE(4f)单元位点,更靠近费米面的具有更高能量水平的f带可能作为电子补偿的缓冲带,通过保留LHB来储存TM-O键(图1c,右侧)。电子缓冲f带的形成可能源于:i)RE-O键中离子性质偏好RE位点向配位的O位点电子捐赠,因为RE物种对氧物种具有亲和力;ii)f轨道本身的更高能级和RE物种易于进行价电子捐赠表明更靠近费米面。因此,来自TM-O-RE单元位点梯度轨道耦合的保留的LHB可以在理论上抑制牺牲和过度氧化TM-O共价性,有望为OER提供合适且稳定的活性位点。
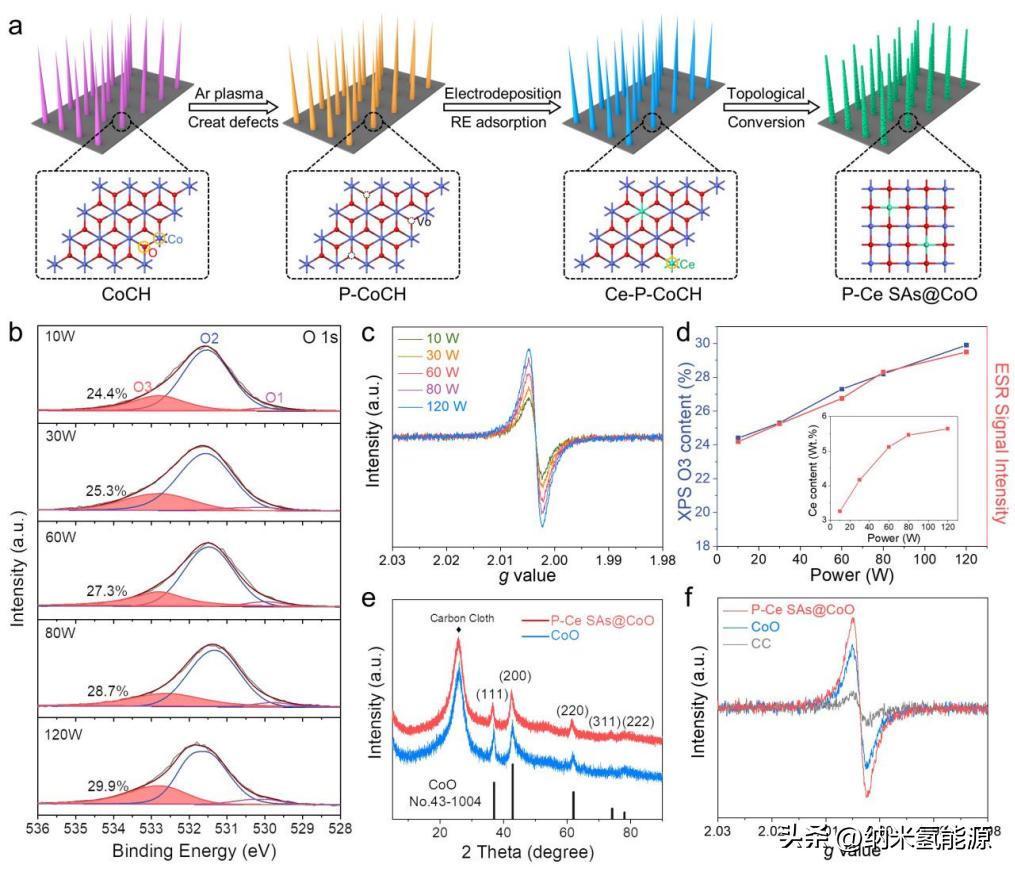
图2 P-Ce的设计策略及物理特性SAs@CoO。(a) P-Ce合成路线示意图SAs@CoO。(b)不同Ar等离子体射频功率下P-CoCH的o1s XPS光谱和(c) ESR光谱。(d) Ar等离子体射频功率与XPS O3含量/EPR信号强度的强度关系。插图为Ar等离子体射频功率与P-Ce中ICP Ce含量的强度关系SAs@CoO;(e) P-Ce SAs@CoO和CoO的XRD谱。(f) P-Ce SAs@CoO、CoO和空白CC的ESR谱。
为了验证上述理论假设,我们提出了一种新颖高效的等离子体(P)辅助策略来构建单原子分散的Ce在CoO表面(P-Ce SAs@CoO),并将其作为一个模型来探索4f-2p-3d梯度轨道耦合对OER的影响。图2a描述了P-Ce SAs@CoO的合成路线。在合成中,由不同射频功率的Ar等离子体处理引起的氧空位(Vo)的形成,在Ce SAs的高效锚定和原子分散中起着至关重要的作用。X射线光电子能谱(XPS)表明,Vo在CoCH前驱物中的比例从24.4%增加到29.9%随着RF功率的增加而增加(图2b)。这个结果进一步被电子自旋共振(ESR)所证实,在其中Vo的ESR未配对电子在g=2.004处的信号随着RF功率的增强而显著增强(图2c)。 XPS分析的Vo含量和ESR信号强度都显示出与Ar等离子体强度的变化良好的一致性,Ar等离子体倾向于随着RF功率的增强呈线性增加(图2d),证明Ar等离子体可以有效地修改催化剂表面以产生氧空位。通过感应耦合等离子体质谱(ICP-MS)定量评估了P-Ce SAs@CoO中不同Ar等离子体RF功率的Ce含量;P-Ce SAs@CoO中的Ce含量从低RF功率的3.26%迅速增加到>5%,并随着功率或Vo含量的增加而饱和在~5.64%(图2d,插图)。组成和结构表征(图S3)表明Ce的含量不会改变CoO的主晶相和结构。这些结果表明,此外,引入Ce后,P-Ce SAs@ CoO的Co 2p 3/2 的结合能相对于CoO负移≈0.50 eV,表明Ce和CoO之间存在强烈的电子相互作用。由于Ce和Co元素之间的原子半径,配位数和价态的差异,原位掺杂的Ce在CoO中易于驱动晶格氧的畸变/位移,从而导致CoO具有Vo丰富的特性。如图2f所示,引入Ce后P-Ce SAs@ CoO的ESR信号显著增加。掺杂了杂质原子Ce的CoO不仅产生更多的催化活性位点,而且通过面共享的Ce-O-Co几何结构优化了Co位点的局部电荷分布(图S6),使得CoO成为一个优化的几何结构和调整的d带配置。
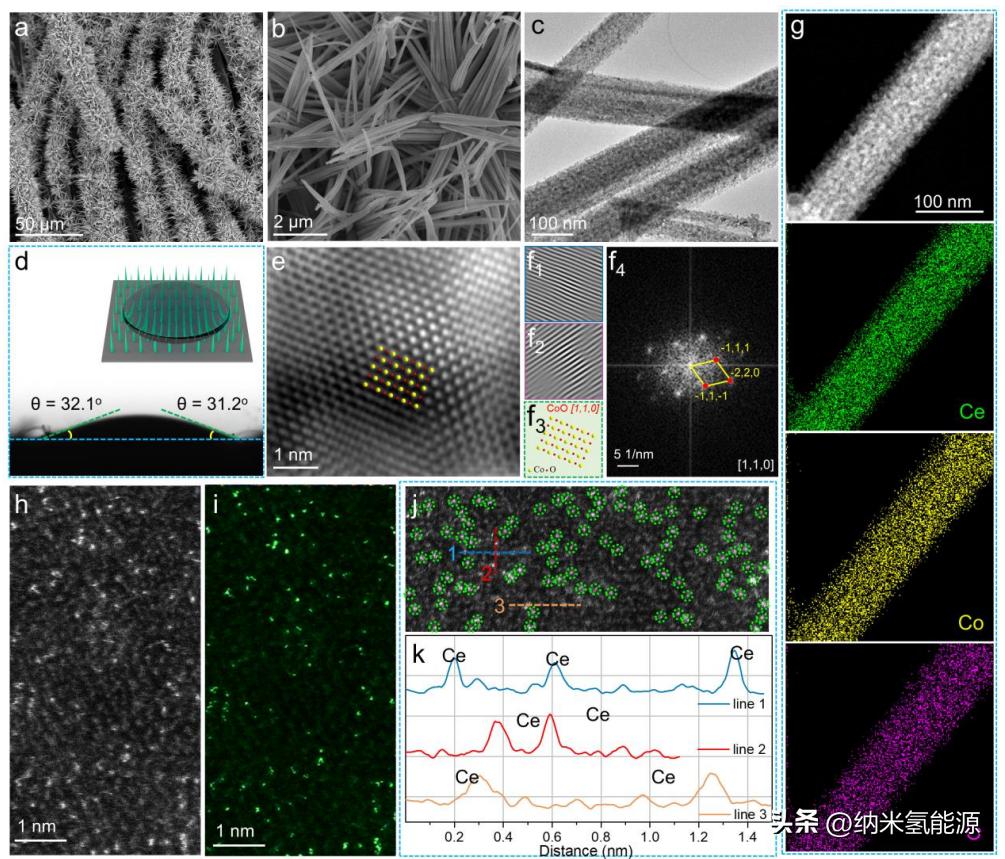
图3 P-Ce的形态和结构表征SAs@CoO。(a、b) SEM图像;(c)透射电镜图像;(d)水滴接触角试验;(e) HRTEM图像;(f1-f2) FFT提取的moir<s:1>图像,(f3) CoO的原子排列模型[110],(f4) e对应的SAED图像;(g) HADDF图像和相应的要素测绘概况;(h-j) AC HADDF-STEM图像;(k) P-Ce SAs@CoO的像素强度积分,分别取自图j中的蓝、红、黄线。
在图3中,我们对P-Ce SAs@CoO的微观/纳米结构以及Ce SAs的分散进行了表征。扫描电子显微镜(SEM)显示,P-Ce SAs@ CoO仍保留了CoCH的初始1D纳米针结构(图3a-b),除了纳米针的光滑表面变得粗糙和多孔。透射电子显微镜(TEM)揭示了纳米针的内部结构由松散连接的颗粒组成(图3c)。这种1D多孔纳米针结构通常提供高比表面积、易于访问的活性位点、丰富的电解质/催化剂界面和快速的质量/电子扩散路径,有利于OER性能。接触角测量(图3d和附图S7)表明,在引入Ce后,P-Ce SAs@CoO的亲水性得到了优化。显著的亲水性可以加速电解液和电极之间的电荷转移速率,从而提高OER活性。P-Ce SAs@CoO的高分辨率TEM(HRTEM)图像显示了周期有序的原子排列(图3e),与模拟的CoO原子排列模型相一致(图3f3)。此外,从HRTEM中没有发现Ce或CeO2颗粒。能谱显微镜(EDS)元素映射图像(图3g)和线扫描剖面(附图S9)证明了Ce在P-Ce SAs@CoO中的成功引入,与ICP-MS结果一致。像差校正(AC)高角度环形暗场扫描TEM(HAADF-STEM)图像清楚地验证了单原子Ce位点在CoO中的原子分散(图3h-j)。此外,亮度强度相关线(图3j)显示Ce原子位于同一列中的Co原子旁边,表明Ce取代了CoO晶格中的八面体配位-Co。因此,可以认为Ar等离子体处理方法可以有效诱导大半径Ce原子在CoO中的锚定和原子分散,协助构建梯度4f-2p-3d轨道耦合的Ce-O-Co几何结构。为了比较,我们还通过类似的方法合成了不含Ce的CoO(图S10)和无等离子体处理的Ce-CoO(图S11)对照物。
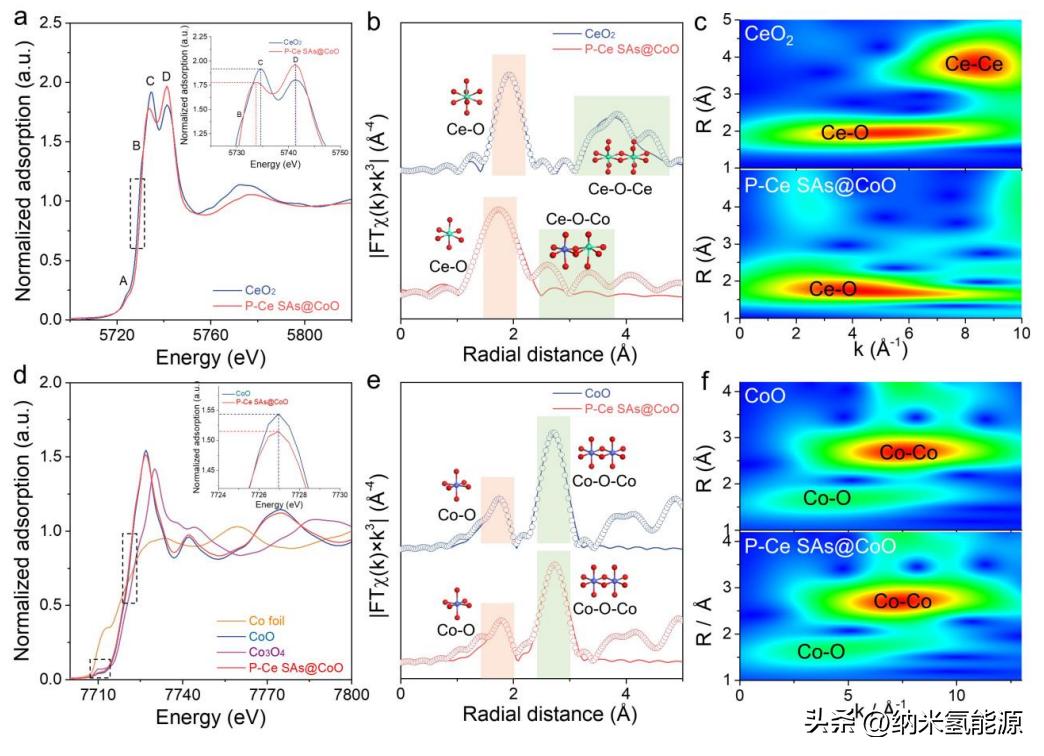
图4 Ce和Co的化学态和配位环境(a)归一化Ce l3边缘XANES光谱;(b) k3权重Ce l3边缘EXAFS谱的傅里叶变换及CeO2和P-Ce的拟合结果SAs@CoO;(c) CeO2和P-Ce的k3权值Ce l3边缘EXAFS信号的小波变换SAs@CoO;(d)归一化Co k边XANES光谱;(e) k3权值Co k边EXAFS谱的傅里叶变换及CoO和P-Ce的拟合结果SAs@CoO;(f) CoO和P-Ce SAs@CoO的k3权值Ce l3边缘EXAFS信号的小波变换。
为了更好地说明P-Ce SAs@CoO中Ce-O-Co位点的化学状态和电子构型特征,使用X射线吸收精细结构(XAFS)表征方法。首先,通过Ce L3边缘X射线吸收近边结构(XAENS)光谱对比表征了P-Ce SAs@CoO和标准CeO2中Ce位点的化学状态。如图4a所示,对于P-Ce SAs@CoO中的Ce L3边缘XANES光谱,白线峰位于更高的5741.3 eV处,而C峰则收缩到较低的5734.5 eV处,表明Ce4+的2p→(4f_0)5d电子跃迁起着主导作用。同样地,与标准CeO2相比,P-Ce SAs@CoO的Ce L3吸收边也逐渐向高能侧移动,说明P-Ce SAs@CoO中Ce 4f轨道状态更空虚,这一点可以通过Ce L3边缘XAENS光谱的二阶导数偏离趋势得到验证(图S12)。Ce 4f轨道状态的这种降低必然会通过Ce-O-Co单元链向周围的Co-O提供电子,从而进一步增强Co-O共价性,并抑制OER过程中Co-O的电子损失。此外,P-Ce SAs@CoO和CeO2之间的这种电子状态不一致现象,主要是由于Ce原子在CoO相中的限制以及局部Co-O-Ce-O-Co螯合结构造成的。为了评估Co-O-Ce-O-Co链式环境对本地Ce-O键合特征的影响,引入了扩展X射线吸收精细结构(EXAFS)技术(图4b和图S14)。未校正相位的傅里叶变换EXAFS(FT-EXAFS)结果(图4b)显示,相对于CeO2(1.90 Å),P-Ce SAs@CoO的Ce-O键长(1.77 Å)略微缩短了约7%,表明了Ce-O-Co单元链中Ce和O之间的共价收缩。此外,这种Ce-O键缩短现象归因于P-Ce SAs@CoO和CeO2晶体结构中Ce-O-Co和Ce-O-Ce之间配位差异的影响,证实了P-Ce SAs@CoO中Ce-O-M键的峰弱化/衰减现象(约4 Å)。EXAFS小波变换分析(图4c)也表明P-Ce SAs@CoO中9 Å^-1处Ce-O-Ce配位信号的消失和弱化,进一步验证了在P-Ce SAs@CoO中与CeO2的第二壳层的不同性质。这意味着,相比于CeO2,CoO为Ce原子提供了完全不同的配位环境,在CoO相中的Ce容纳导致调节了Ce-O键合强度并形成了Ce单原子位点,这倾向于影响OER过程中的中间体吸附。
对比于明显调制的Ce,P-Ce SAs@CoO中的宿主CoO显示出与CoO样品相似的化学状态和配位环境。图4d展示了P-Ce SAs@CoO、CoO、Co3O4和Co箔片的Co K边X射线吸收近边结构(XANES)光谱。P-Ce SAs@CoO的Co K边吸收曲线与CoO几乎重合,表明P-Ce SAs@CoO中的Co元素大多以Co2+的形式存在。由于高度对称的六角八面体[CoO6]单元中1s到3d的偶极禁戒电子跃迁,P-Ce SAs@CoO的预边峰与Co3O4相比变得更弱甚至消失,表明引入Ce不改变CoO的批量相结构。此外,在P-Ce SAs@CoO和CoO的Co K边XANES轮廓进行仔细比较后,发现随着构建Ce单原子位点,吸收边向低能量侧移,表明Co的价态略微降低,这可能是由Ce的电子供应引起的。此外,Co K边白线强度在引入Ce后显示出微弱的趋势,进一步验证了Co氧化态的降低。通过扩展X射线吸收精细结构(EXAFS)进一步研究了P-Ce SAs@CoO中Co的配位配置(图4e和图S15-16)。如图4e所示,P-Ce SAs@CoO和CoO的相位未校正的FT-EXAFS光谱的主要峰在第一壳层区域都表现出类似的1.75 Å的峰(归因于Co-O路径)。第二壳层区域的强Co-O-Co峰(峰位于2.71 Å处)的存在进一步证实了P-Ce SAs@CoO中宿主CoO相的形成。EXAFS小波变换分析(图4f)表明,P-Ce SAs@CoO和CoO均表现出两个高强度峰,分别位于k = 4和8 Å,分别对应于Co-O和Co-O-Co路径,表明负载的Ce原子不会改变CoO的体相配位配置。因此,可以得出结论,构建Ce-O-Co单元链显著缩短了Ce原子点处的Ce-O键长,捐赠了4f电子并导致了P-Ce SAs@CoO中Co氧化态的降低。
电化学测试
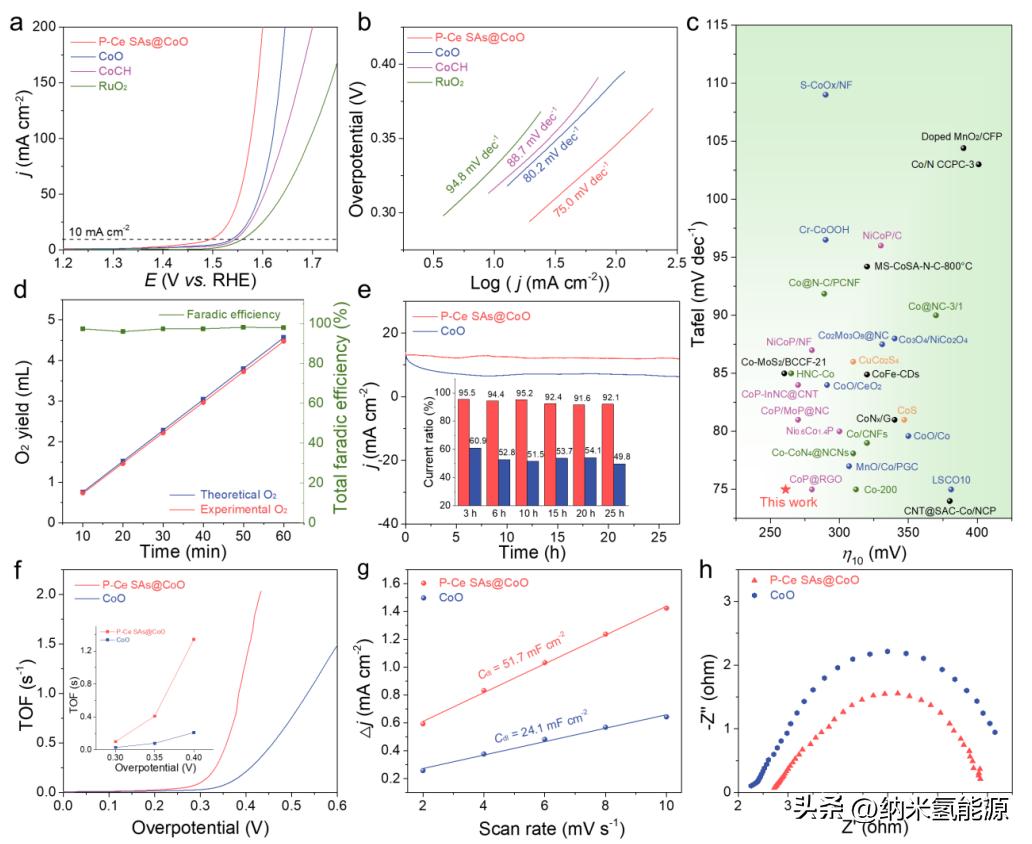
图5 电解OER性能。P-Ce SAs@CoO及其它同类材料在o2饱和1.0 M KOH溶液中的电化学OER性能:(a) LSV曲线;(b)根据(a)得出的塔菲尔图;(c) OER性能与以前报道的其他钴基催化剂的比较;(d)法拉第效率试验;(e)通过计时电流测试进行稳定性测试(插入显示保留电流的百分比);(f) TOF图,插图显示了不同过电位下的TOF值;(g)不同扫描速率下CV图的Cdl值;(h) EIS奈奎斯特图。
P-Ce SAs@CoO是一种在CoO中原子分散的单个Ce位点催化剂,对于研究Ce-O-Co活性位点对OER的影响提供了机会。通过iR-校正线性扫描伏安法(LSV)在饱和O2的1.0 M KOH溶液中执行不同Ce含量的P-Ce SAs@CoO的OER极化曲线来得到最佳催化剂。中等Ce含量为5.46 Wt.%的P-Ce SAs@CoO表现出最佳的OER活性,具有较低的过电势和快速的电流上升。优化后的P-Ce SAs@CoO与其他材料的OER极化曲线进行比较,发现其在整个电势范围内显示出更高的电流响应,并且甚至比商业RuO2催化剂更高。P-Ce SAs@CoO在10mA cm−2的过电势仅为261 mV,比CoO和RuO2分别降低了49和71 mV。此外,与P-Ce SAs@CoO相比,无Ce的CoO需要更高的能量屏障。Tafel图表明,在多电子转移路径中,P-Ce SAs@CoO表现出更容易的OER动力学,且速率决定性步骤倾向于位于M-O形成步骤,证明构造Ce-O-Co活性位点可以加速电子反应。稳定性测试表明,制备好的P-Ce SAs@CoO在经历过时间的恒流测试后仍保持着很好的稳定性。P-Ce SAs@CoO的基本结构单元和组成在稳定性测试后仍能保持良好。总的来说,P-Ce SAs@CoO在OER方面的优异表现和电化学稳定性使其具有潜在的实际应用价值,这在整体水分解装置中得到了证明。
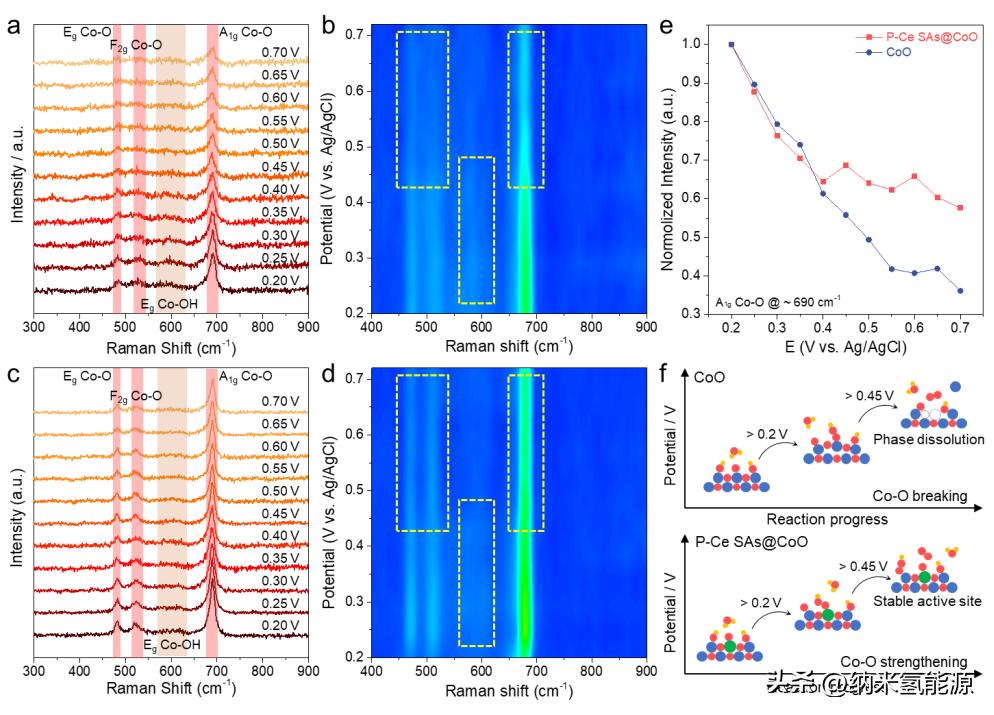
图6 原位拉曼光谱研究P-Ce OER机理SAs@CoO。(a)原位拉曼光谱和(b) CoO对应等值线图;(c)原位拉曼光谱和(d) P-Ce对应等高线图SAs@CoO;(e) 690 cm-1处归一化拉曼峰;(f) P-Ce SAs@CoO和CoO的OER表面演化过程示意图比较。
为了获取Ce-O-Co单元对OER机理的影响,采用电化学原位拉曼光谱监测P-Ce SAs@CoO和CoO在OER条件下的表面变化。CoO(图6a)和P-Ce SAs@CoO(图6c)的拉曼光谱显示了一组峰,包括分别对应于Co-O相中Eg、F2g和A1g振动模式的482、521和690 cm-1三个典型特征峰。在低偏压电位(0.2-0.45 V vs. Ag/AgCl)下,580-620 cm-1范围内的宽峰被归属为CoO表面吸附OH-物种的Eg振动模式,这可能是由于1.0 M KOH溶液中CoO表面吸附了OH-离子引起的。研究了这些特征峰强度变化在不同偏压电位梯度下的情况,以了解P-Ce SAs@CoO和CoO表面在OER过程中的转化情况。图6b和图6d分别展示了CoO和P-Ce SAs@CoO的拉曼光谱轮廓图。在0.2 V vs. Ag/AgCl的工作偏压电位下,CoO呈现出良好信号强度的Co-O Eg、F2g和A1g振动峰。随着偏压电位的增加到约0.45 V vs. Ag/AgCl,Co-O振动峰的信号强度表现出明显的快速衰减趋势,这可能是由于OER过程中催化剂的表面重构、晶格氧演化和溶解-沉积作用所致。当偏压电位持续增加到0.45 V vs. Ag/AgCl以上时,Co-O振动峰的信号强度继续衰减,甚至在轮廓图中模糊不清。OER过程中的逆境条件,包括强碱性和氧化环境,通常会触发催化剂的溶解。然而,在引入Ce SAs后,P-Ce SAs@CoO即使在高达0.45 V vs. Ag/AgCl的高偏压下仍保留了稳固的Co-O键,显示出良好的Eg、F2g和A1g Co-O振动峰,证明了P-Ce SAs@CoO的稳定性。此外,Figure 6d中的轮廓图进一步证实了Ce的保护效果。因此,Co-O A1g(690 cm-1处)在P-Ce SAs@CoO和CoO的标准化残余峰强度的变化(图6e)进一步支持了稳定的OER活性Ce-O-Co单元的成功构建,该单元在严酷的OER条件下可以存活。总的来说,在CoO中构造Ce-O-Co单元打破了原始的Co-O键的溶解/断裂趋势,而Ce则保护了Co-O位点,并通过Ce引起的电子重构提供了稳定和高效的OER单元(图6f)。
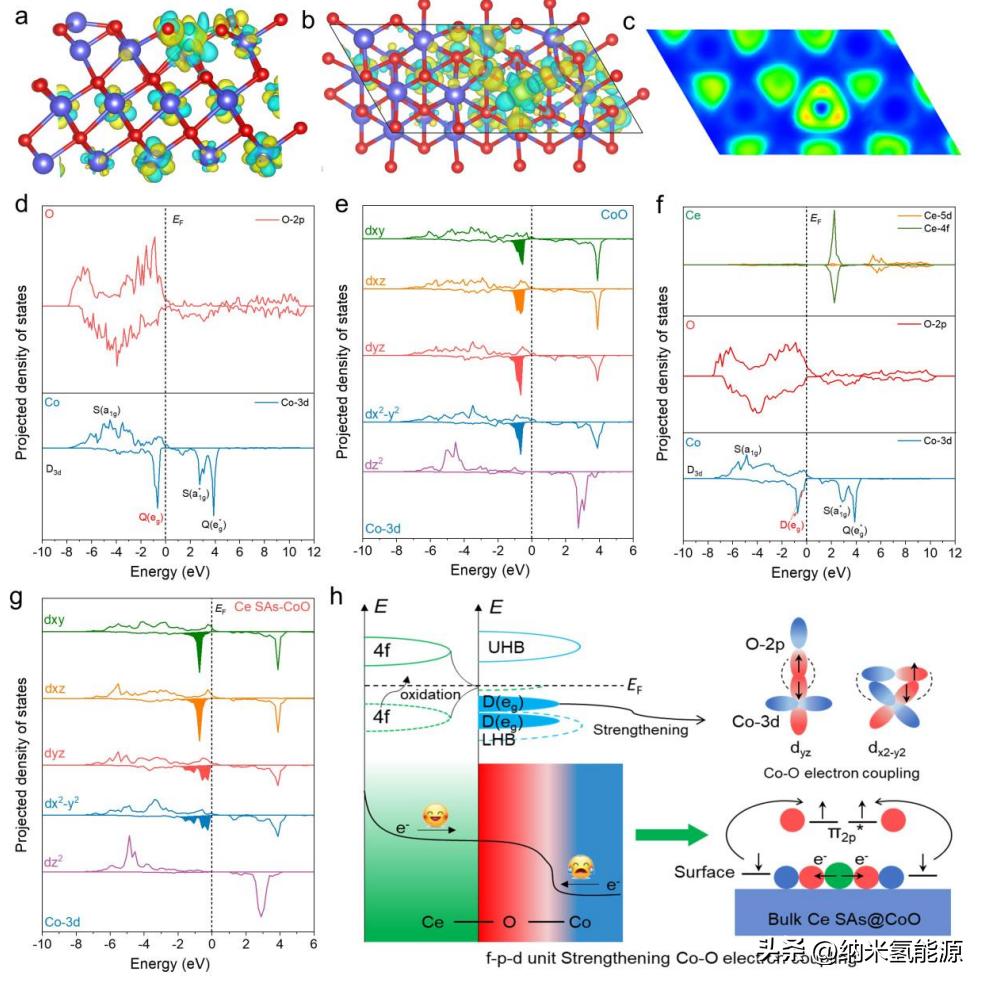
图7 Ce-CoO的电子结构分析。电荷密度差:侧面(a)和(b)俯视图。黄色区域表示电子积累,蓝绿色区域表示电子消耗。配色:蓝色为Co,红色为O,黄色为Ce;(c) Ce-CoO的ELF;(d) Co-3d和O-2p在CoO(111)中的PDOS与(e) Co-3d的详细投影状态;(f) Ce6c-O-Co6c单元位上Ce-4f、O-2p和Co-3d的PDOS与(g) Co-3d相应的轨道投影态;(h)通过Mott-Hubbard模型和OER的自旋选择耦合,提出Ce6c在Ce6c- o - co6c单元位点的电子调制效应。配色方案:蓝色代表Co,绿色代表Ce,红色代表O。
针对CoO中的单原子Ce所做的自旋极化DFT+U计算的结果。通过将体块CoO结构(空间群:Fm3̅m)剖离成CoO(111)表面,可以将体块中的6配位Co6c位点转化为表面Co6c位点。因此,单原子Ce更倾向于占据6配位的Ce6c位点。通过电荷密度差异分析发现,与周围的Co6c位点相比,单原子Ce共享了更多的电子密度,而在Ce-O键处O仍然充当电子受体。通过电子局域化函数(ELF)分析,发现Ce6c位点向配位的O供体了更多的电子,并赋予了Ce-O键离子性质。通过CoO的投影态密度(PDOS)图谱发现,由于Co6c的局部对称性(D3d),Co-3d态可以被分类为单一的a1g和四重的eg。通过Ce-4f的调制效应,eg态在两个双重退化的D(eg)态上进一步轨道分裂,其中自旋下的dyz和dx2-y2态对EF周围的电子分布做出了更大的贡献。这种轨道分裂可以解释由Ce6c-O-Co6c单元位点中Ce-4f态的电子捐赠引起的Co-3d的自旋选择性固定氧物种电子的激活,这对OER过程非常重要。通过将Ce6c纳入构造局部的Ce6c-O-Co6c单元位点,Ce-4f态的电子捐赠可抵消LHB的电子损失,保持M-O的稳定性,并同时通过晶格应变对称性的破坏,将D(eg)态的自旋下电子推向EF更近。对于O2的三重基态(π2p*↑↑),自旋下的dyz和dx2-y2轨道可与氧物种形成σ和π键相互作用,提供了自旋选择性固定效果,以指导OER过程中O2的生成。

图8 OER活性和稳定性的理论分析。(a)不同*OH覆盖率的CoO(111)优化模型;(b) 25℃、pH =14时计算的表面Pourbaix图;(c) 10 mA cm-2的动力学OER活火山模型,函数为G*O-G*OH;(d)火山模型预测的实验电位和理论电位的比较。(e)在* oh覆盖的CoO和Ce-CoO(111)上的OER路径,图中有Bader电荷族;(f) CoO和Ce在1.23 V vs. RHE偏置电位下的Gibbs自由能图CoO(111);(g) CoO与Ce-Co的标度关系分析(111)。
本文进一步分析了Ce-CoO在OER中的活性和稳定性,通过表面Pourbaix图和高级微观动力学模型推导出理论OER电位。考虑到CoO(111)上OH覆盖度,计算了1/8到7/8 ML OH范围内所有可能的吸附位点(图8a)。根据计算得到的表面Pourbaix图(图8b),在pH=14时URHE >1.40 V时,CoO(111)可以通过7/8 ML OH稳定。基于有利的7/8 ML OH覆盖CoO,在OER操作条件下,使用高级OER微观动力学模型在10 mA cm-2时考虑了所有基元反应的动力学和热力学,以GO-G OH作为有效OER描述符来分析OER活性(图8c和图S25)。可以看出,许多掺杂Ce的位点具有出色的OER性能,活性集中于火山峰,并优于CoO(111)。绘制10 mA cm-2时预测与实验电位的对比图,发现理论计算与实验之间有极好的一致性(图8d)。在电化学有利的OH覆盖表面吸附OH后,随后加入H+/e-对会将OH转化为O和OOH, 分别形成各自的结构。CoO和Ce-CoO(111)具有OH覆盖效应的不同结构显示在图S26-27中,并且显示出活性中心从Co6c-O-Co6c转变为Ce6c-O-Co6c。引入Ce到CoO(111)中会导致表面电荷重分布,Ce位点作为电子给体,其波德电荷(+2.31 e)高于Co(+1.51 e)(图8e)。由于电子给体效应的存在,Co位点的自氧化得到缓解,以保持局部电子密度,并通过降低Co位点的波德电荷,这一点得到了证实,由+1.51 e降至+1.26 e。对于CoO(111),最大能隙是将O转变为OOH,其能量差为0.498 eV(也称为限制电位步骤,PDS)。而对于Ce6c-O-Co6c单元位点,Ce-CoO(111)的PDS变为将OH转变为O,能量差减小到0.417 eV(图8f),显示出减少了热力学限制。由于这两个PDS都处于从OH向OOH的转变区域内,进一步分析了ΔG(*OOH)和ΔG(*OH)之间的标度关系。因为ΔG(*OOH)和ΔG(*OH)之间的最佳能隙是2.46 eV,接近2.46 eV的值表明更高的OER活性[55,56]。如图8g所示,CoO(111)的标度关系ΔG(*OOH)-ΔG(*OH)为3.3 eV,而对于Ce-CoO(111),独特的电子调制效应将ΔG(*OOH)-ΔG(*OH)从3.3升高到3.25 eV,增强了OER性能。
结论
本文通过一种新颖的等离子体辅助策略(P-Ce SAs@CoO)将单原子Ce引入到CoO中,开发了一种高性能的OER催化剂。与CoO和基准材料RuO2相比,P-Ce SAs@CoO表现出优异的OER性能,其超低过电位为261 mV。通过离线XAS、原位拉曼、DFT、表面状态分析和微观动力学建模,阐明了活性位点并提供了机理上的见解。结果表明,Ce(4f)-O(2p)-Co(3d)的梯度轨道耦合赋予Ce-O-Co单元位点高效的OER活性。这种独特的Ce(4f)-O(2p)-Co(3d)的梯度轨道耦合加强了Co(3d)-O(2p)的共价性,从而增强了OER性能。O和OH的平衡吸附使Ce-CoO的GO-GOH更接近于微观动力学火山模型的顶点。本研究不仅建立了一种高性能的*土稀**基OER催化剂,还提供了有效的f-p-d梯度轨道耦合策略,可以设计出更活性和稳定的*土稀**-过渡金属氧化物(RE-TMO)催化剂用于电催化。
参考文献
Li, M., Wang, X., Liu, K., Sun, H., Sun, D., Huang, K., Tang, Y., Xing, W., Li, H. and Fu, G. (2023), Reinforcing CoO Covalency via Ce(4f)─O(2p)─Co(3d) Gradient Orbital Coupling for High-Efficiency Oxygen Evolution. Adv. Mater. 2302462.