文|渊溪的竹简
编辑|渊溪的竹简
前言
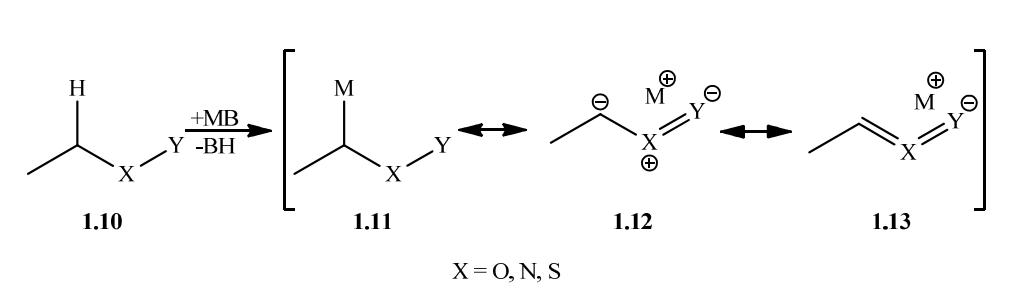
静态偶联是一种重要的有机合成反应,通过在钯催化下将有机锡化合物与有机卤化物或伪卤化物进行偶联反应,可以高效地构建碳-碳键,在这个过程中,手性甲基钯配合物扮演着关键的角色,其构型稳定性对于控制反应的立体选择性和产物的手性纯度具有重要影响。
而手性甲基钯配合物就是是由手性配体与钯离子形成的配位化合物,这些手性配体通常是基于膦、膦酸或其他手性配体的衍生物,手性配体的选择和设计直接决定了钯配合物的手性构型,在静态偶联反应中,手性甲基钯配合物作为催化剂参与反应,促使有机锡化合物与有机卤化物之间形成新的碳-碳键。
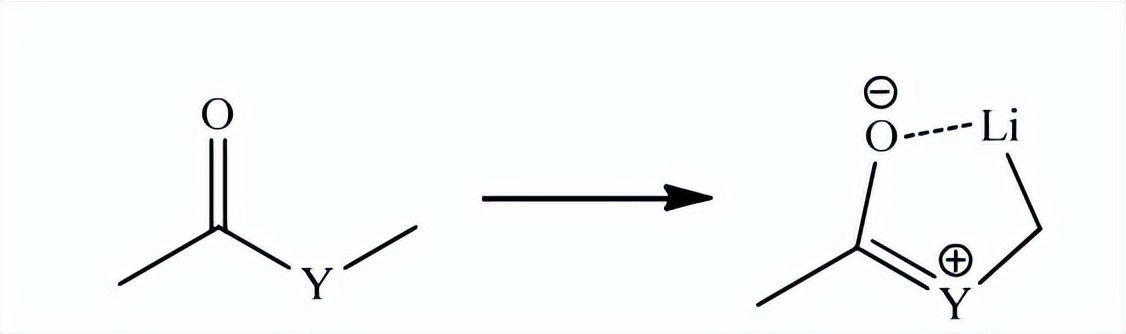
三丁基苯乙烯基甲醇的偶联-手性羟甲基钯配合物
首先,我们希望研究通过斯蒂勒偶联直接转化溴苯和三丁基基甲醇形成的羟甲基钯配合物2.31的微观构型稳定性。
在我们的研究小组中,不需要对三丁基基甲醇进行衍生化处理,因为它是一个众所周知的前体物,这是因为它用于偶联反应,由于日本研究人员在1985年已经进行了与三丁基基甲醇的斯蒂勒反应。
因此我可以简单地重复这些实验,使用三丁基基甲醇和(S)-三丁基基-[D1]甲醇在80°C下反应18小时。
偶联后唯一必要的步骤是将标记的苯基甲醇2.32与(S)-MTPACl酯化,以确定其构型和对映体过量,我们记录过(R)-Moser酯的1H NMR光谱,它们作为参考光谱被使用,在1H NMR光谱中显示出CHD基团的部分显示,由(S)-三丁基基-[D1]甲醇衍生的苯基-[D1]甲醇是(R)-构型的,并且具有99%的对应异构体过量。
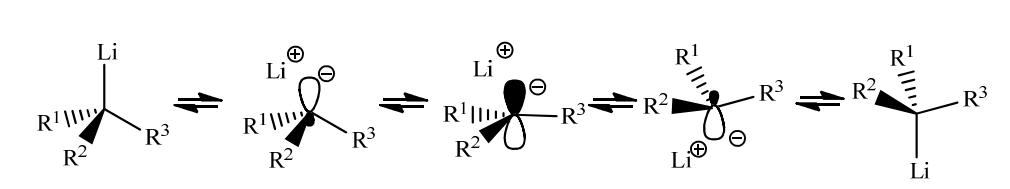
所以说,静态偶联反应以构型的净保持进行,因此,金属转移和还原消除过程必须以保持或倒转的方式进行,由于(R)-Mosher酯下面的苯基-[D1]甲醇的对映异构体过量大于99%,在斯蒂勒偶联反应中形成的各种钯配合物在还原消除产物之前在微观上是构型稳定的。
还有在我们实验工作的这部分中,我们希望研究在静态偶联反应过程中由(R)-或(S)-1-三丁基基-[D1]甲基苯甲酸酯和苯甲酰氯生成的钯配合物2.34的微观构型稳定性。
我们首先使用未标记的物种进行了所有的反应,以确定它们是否可以以合理的产率进行,并观察是否有可能通过光谱方法确定产物的构型和对映异构体过量。
为了进行偶联反应,我需要通过将三丁基基甲醇(1.67)与苯甲酰氯进行简单的酯化反应制备前体2.35,而且在偶联反应中,我们还使用了Stille等人报道的方法,用于将α-三丁基基甲酸酯与苯甲酰氯进行偶联。
由于无法直接通过光谱方法确定该反应产物的对应异构体过量,因此需要进行另外两步反应转化。
得到的苯甲酸酯2.36经过LiALH4的还原反应,生成了一个不标记系列中的一个拉吡司二醇2.37的混合物,以及一个标记系列中的两个非对映异构体的二醇混合物,以及源自苯甲酰基的苯甲醇,标记的碳原子的构型没有改变。
最后一步是使用(S)-MTPACl对二醇进行酯化反应,得到两个非对映异构体的(R)-Mosher酯2.38,这只针对标记的物种进行,因为已知未标记酯的参考光谱。
通过与Kapeller记录的参考光谱进行比较,可以确定酯的标记碳原子的绝对构型和对映异构体过量,由于偶联之前和之后的所有步骤都是保持构型的,我们证明了偶联反应以构型的净保持进行。
这意味着金属转移和还原消除过程都可以通过保持构型或倒转构型进行,但是保持构型的机制更为可行,进一步证明,由(R)-或(S)-三丁基基-[D1]甲基苯甲酸酯和苯甲酰氯生成的钯配合物2.34在70°C下。
在斯蒂勒偶联反应过程中在微观上是构型稳定的,因为每种情况下标记的碳原子的对映异构体过量都等于或大于99%,在这里,静态偶联反应必须经过一个闭合的过渡态2.39,以解释偶联步骤的构型净保持现象。
手性硫甲基钯配合物的微观构型稳定性
在我们的实验工作中,最大的挑战是制备和确定含硫的各种钯配合物的构型稳定性。
原则上,一切都应该很顺利,并且在偶联后进行的步骤以确定对映异构体过量已经被充分确立,通过相关的方法,使用由苯基甲硫醇和α-溴代苯乙酮制备的未标记化合物2.42a进行了用于确定标记的α-苄基硫乙酮的对映异构体过量的初步实验。
在干燥的THF中,使用LiAlH4进行还原反应,产生了所需的醇2.43,收率为96%,虽然没有进行(S)-MTPACl的酯化反应,但预计会生成一系列非对应异构体的Mosher酯混合物2.44。
在我们尝试对未标记的(苄硫甲基)三丁基锡烷(2.45a)进行斯蒂勒偶联反应时,随后是对标记物种的反应,因为我们用完了未标记物质,包括苯甲酰氯或溴苯,最后,我们用乙酰基取代了锡烷中的苄基作为保护基团。
另外,我们尝试了所知的各种溶剂、Pd催化剂和配体,然而,这些试验并未针对含有硫原子的化合物,在需要进行偶联的碳原子的α-位置。
但是所期望的产物从未形成,只有在第一次实验中,起始物质被消耗掉,只不过没有检测到偶联产物,看起来产物形成了,但在这些反应条件下发生了自缩合反应。
因此,从室温开始重复进行斯蒂勒偶联反应,反应的进展通过薄层层析法(TLC)进行监测,并且参考样品2.42a也被涂抹在薄层板上,不幸的是,没有形成偶联产物,起始物质在这次实验中一点都没有消失,尽管温度提高到与前一次实验相同的温度。
在那个时候,决定尝试使用不同的催化剂对溴苯进行偶联反应,以查看是否能得到所期望的产物2.47a。
接下来我们尝试的条件是添加过量的氟化铯(CsF)(实验5),这是非常有希望的条件,因为它们在许多不活泼的有机锡试剂和芳香卤化物上都取得了成功,这次形成了一个产物,乍一看看起来很不错,但在1H NMR光谱中的积分显示,它并不是所期望的二硫化物2.47a,而是显然由硫-碳偶联反应形成的产物。
有可能氟化物添加剂导致苯甲硫醇的形成,并与溴苯发生偶联反应,因此我们尝试了没有氟化铯的反应,但不幸的是这也没有得到所期望的产物。
我们决定再给一次含有乙酰保护基的硫甲基锡烷进行静态偶联反应的最后两次尝试,前体是通过Mitsunobu反应由三丁基锡甲醇和硫乙酸制备而成的。
然而,即使是这个前驱体也不能与溴苯偶联,它的反应性不足以被消耗掉。
偶联-手性氨基甲基钯配合物的微观构型稳定性
最后,我们研究了由(R)-N-三丁基锡基-[D1]甲基邻酰苯酰亚胺和苯甲酰氯得到的N-保护氨甲基钯配合物的微观构型稳定性。
和其他情况一样,通过简单的Mitsunobu反应(49)制备了标记和未标记的前体,反应中使用未标记和标记的三丁基锡基甲醇(1.67)和邻酰苯酰亚胺,反应过程中发生了构型的倒转(SN2反应)。
静态反应使用的条件与与苯甲酰氯进行偶联的类似底物相同,在存在CuCN的条件下进行的反应收率几乎相同,标记和未标记底物的收率分别为77%和78%,反应通过TLC监测,为了确定标记产物的对映体过量,需要进行两个简单的步骤。
酮2.55经过Parr装置中大量催化剂的催化还原生成醇2.56(51),尽管收率的重现性较低,但即使是较低的收率也是可以接受的,因为只需要约10毫克的产物就足够进行下一步的酯化反应,即与(S)-MTPACl酯化反应,得到二对映异构体的混合物,通过1H NMR光谱测定,对映体过量大于98%。
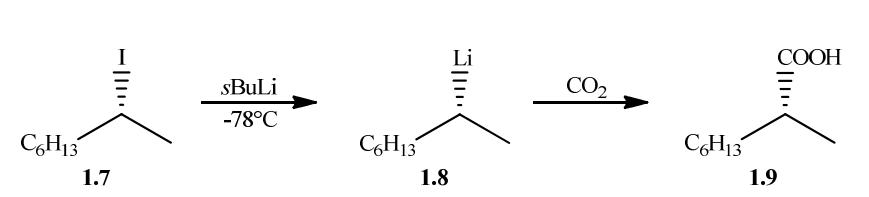
因此,Stille偶联反应中形成的中间体钯配合物2.53,由Pd0配合物、(R)-N-三丁基锡基-[D1]甲基邻酰苯酰亚胺和苯甲酰氯形成,在75°C条件下微观上具有构型稳定性,由于没有可用的参考样品,无法确定氮上的绝对构型。
为了确定绝对构型,我决定使用类似的钯配合物2.58,假设与苯甲酰氯和溴苯甲烷的Stille偶联反应遵循相同的立体化学过程。
这些配合物是由未标记和标记的2.54与溴苯甲烷生成,用于制备邻酰苯酰亚胺2.59,这次的Stille偶联反应比与苯甲酰氯的反应要慢得多,尽管在未标记系列中产率较低,但在标记系列中使用了氨基-[D1]甲基锡烷的两个对映体,在干烷基苯和二恶烷中进行反应,以研究不同溶剂的影响,通过化学对应,确定了标记产物的构型。
通过进行以对映体反转为特点的Mitsunobu反应,利用(R)-苯基-[D1]甲醇制备了已知构型的参考样品,随后,对N-取代的邻苯二甲酰亚胺进行去保护,并以与Stille偶联产物相同的方法将其转化为(R)-Mosher酰胺。
尝试了不同的条件进行Stille偶联,以找到产率最高的条件,总结起来,与苯甲酰氯的偶联相比,该偶联的效果并不理想,然而,产率仍然足够高,以确定产物的构型和对映体过量。
经过实验发现,对于未标记底物而言,最佳条件是在甲苯或二恶烷中,以Pd(PPh3)2Cl2(8 mol %)和CuCN(16 mol %)为催化剂,在100 °C下进行Stille偶联(第5和第6步),而对于标记的邻苯二酰亚胺2.54的两个对映体,也是在甲苯和二恶烷中进行偶联。
为了确定N-苯基-[D1]甲基邻苯二酰亚胺的绝对构型和其对映体过量,邻苯二酰亚胺2.59经过去保护和转化得到(R)-Mosher酰胺(方案2.23),该序列是通过对未标记的邻苯二酰亚胺2.59进行优化的。
在使用肼水合物去保护胺基后,小量的苯甲胺的分离证明相当棘手,由于胺也可溶于水中,为了去除邻苯二甲酰亚胺和肼水合物的过量,我们尝试了许多次并对文献中已知的工艺进行了修改,一旦我们成功做到了这一点,即使我们不能完全去除肼水合物,转化为(R)-Mosher酰胺2.62仍然很容易进行。
根据(R)-Mosher酰胺2.62的1H NMR谱与由苯甲胺和(R)-苯基-[D1]甲醇得到的酰胺的谱进行比较,这两个步骤都没有改变绝对构型,结果发现,通过Stille偶联得到的(R)-Mosher酰胺是一种二面体异构体的混合物(底物胺的对映异构体过量为52%),其中主要的异构体具有(S)构型,这意味着Stille偶联主要经历了保留的路径。
类似地,我们假设之前的(R)-N-tributylstannyl-[D1]methylphthalimide (2.54)和苯甲酰氯之间的Stille偶联也完全以保留构型进行。
两者与先前的Stille偶联的区别在于所使用的溶剂以及将苯甲酰氯替换为溴苯,为了弄清部分消旋是否归因于作为溶剂的二氧六环,试验中使用(S)-[D1]2.54邻酰基的苯并二咪唑,以类似于(R)-对映体的方式进行Stille偶联,只是用甲苯替代二氧六环,结果并没有显著不同,偶联主要以保留构型进行,产物的对映异构体过量度增加到69%。
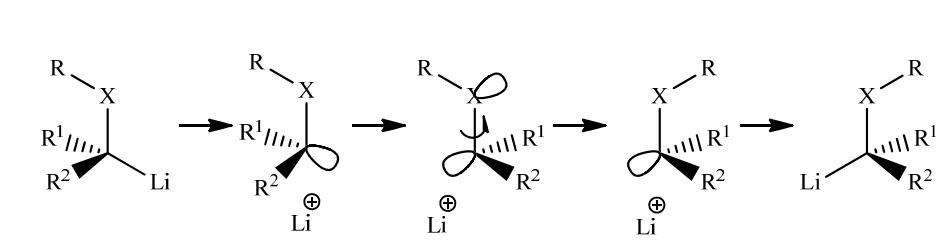
因此,部分对映异构体化的主要因素似乎不是溶剂,剩下两个解释的可能性:要么中间产物的N-保护手性氨甲基钯络合物2.58在显微尺度上具有构型不稳定性,要么金属转移反应通过主要的闭合过渡态2.64(保留构型)和次要的开放过渡态2.63(倒置构型)的组合进行,因此,总体立体化学是构型的保留。
为了查看是否可以通过使用不同的氨基保护基团来改善与亚锡基甲基胺的Stille偶联反应的产率,尝试使用Boc保护基团作为酰胺基团的替代品。
第一个实验没有得到所期望的产物,而是得到了与起始物相同的产物,只是失去了一个丁基基团(实验1),希望这个副反应可以被抑制,实验被重复进行,但是从室温开始,然后将温度升高到50°C,结果基本上与之前的实验相同。
这种相同的副反应在使用邻二酰亚胺保护基进行的Stille偶联反应中也有观察到,但程度要小得多,可能是由于Boc基团中的羰基氧原子配位到锡原子,从而使得丁基基团的转移比保护的氨甲基基团更有利,为什么在邻二酰亚胺基团中观察不到这种情况的原因可能是锡与氧原子的配位比在Boc保护基的前体中不利。
总结
静态偶联过程中手性甲基钯配合物的构型稳定性对于合成手性化合物具有重要意义,这些配合物在适当的反应条件下能够保持其手性构型,从而实现手性信息的传递和保留,然而,构型稳定性可能会受到反应条件的影响,如温度和溶剂选择,合理选择反应条件可以最大程度地确保构型稳定性,并提高手性转移的效率。
另外,对于手性保护基的选择也是至关重要的,不同的保护基可能对钯配合物的构型稳定性和副反应产生不同的影响,因此,对于静态偶联反应中手性甲基钯配合物的构型稳定性的深入研究和优化是合成手性化合物的关键步骤。
参考文献
【1】《高级有机化学,第A部分:结构与机制,第五版》.
【2】《高级有机化学,B部分:反应与合成,第5版》。
【3】《四面体有机化学》。
【4】《在过渡金属催化的不对称转化中具有 C-N 螯合能力的手性配体》。
【5】《与手性配体的对映选择性 Stille 偶联反应》。