MMn既是一系列分子力场的名称,也是一系列分子力学程序(molecularmechanics)的名称。这些分子力场和分子力学程序均由美国乔治亚大学(UniversityofGeorgia)的Allinger教授领导的研究小组开发和发展。作为分子力场的MMn,既是最早被广泛认可,又是最具影响力的分子力场之一。虽然MMn系列分子力场的第一版MM1的势能函数简单、力场参数不多、预报精度不高、应用范围和影响不大。但是,该系列力场的第二版MM2已经相当成熟,预报精度有了很大的提高,在当时的应用范围和影响巨大。当发展到第三版MM3时,已经是一个非常复杂又高度成熟的分子力场。目前,该系列力场的最新版是第四版MM4O与MM3相比,MM4又增加了多项复杂的交叉相互作用,对分子振动频率的预报精度有了较大的提高。因此,MM4仍然是最具影响力、使用最广泛的分子力场之一。
不同于许多主要用于MD模拟的分子力场,MMn系列分子力场的主要应用领域一直是分子力学,其最显著特点包括两个方面:一方面是对分子结构、生成熔、振动频率等的预报精度高;另一方面是适用的分子范围广泛,几乎包括了所有常见的有机分子类型。
4.3.1 原子类型
与其他大多数分子力场一样.MMh系列分子力场根据原子形成化学键时的杂化类型、结构以及与之成键的原子类型(即原子在分子中所处的化学环境)分类原子,确定适用的势函数及其参数。与其他许多分子力场不同的是,MMn系列分子力场利用数字序号表示原子类型,而不是标识符。同时,为了精确描述特定原子在化学环境发生细微区别时势函数的区别,MMn系列分子力场中原子类型较其他类型的分子力场更多。其中,O原子和N原子的类型特别多,但H原子的类型并不是很多。例如,MM2分子力场中只有75种不同的原子类型,但MM3分子力场中则已经增加到149种原子类型。另外,为了保持与前面版本的一致性,MMn系列分子力场的原子类型序号看起来有点零乱,没有任何规律。新版本中增加原子类型时,通常是在原子类型表的后面增加新的类型。常常是前一个版本中的某种原子类型,在后一版本中分出新的类型时,一方面是保留前面的原子类型,另一方面,再在后面增加新的一个原子类型,形成前一版本与后一版本之间的一对多的关系。例如MM3中的第5类氢原子,在MM4中新增了第122类烯怪氢原子类型。有时,前面一个版本的一种类型被取消,那么在后一版本中可能空出一个未用的原子类型。例如,MM2中的20类孤对电子LP类型,在MM3中不再出现,MM3中20类成为未用(notused)的类型。
4.3.2MM" 的势函数形式
为了增加力场所能适用的分子类型,提高对分子的结构、生成熠和振动频率等的预报精度,MMn系列的每个版本多比前一版本引入更多的交叉项、更高次的展开项。早期版本MM1和MM2只有相对简单的势函数,一般不包括交叉项或耦合项,如MM2中只包含一个伸缩-弯曲交叉项。这样的势函数虽然基本不会影响对内部张力较小的大多数分子的构型预报,但对张力较大的分子构型的预报以及振动频率的预报,正确性相对较差。因此,MM3增加到九种类型的势函数,它们分别是键伸缩势、键角弯曲势、二面角扭曲势、离面弯曲势、vanderWaals相互作用势和静电相互作用势,以及键伸缩-弯曲势、二面角扭曲-伸缩势、键角弯曲-弯曲势等交叉项,

在MM4中,又新增加了伸缩-伸缩势、二面角扭曲-键角弯曲势、键角弯曲-扭曲-弯曲势、扭曲-扭曲势、扭曲-贋扭曲势、贋扭曲-扭曲-贋扭曲势等交叉项,同时,MM3中的离面弯曲势被贋扭曲势取代,
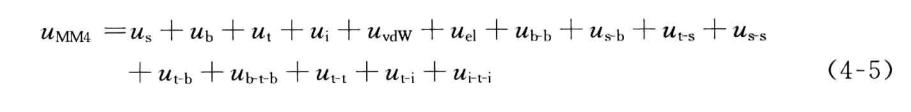
1. 键伸缩势
虽然最常用的共价键伸缩势函数是只包含平方项的谐振子势函数,但是为了提高精度,MMn系列分子力场选择更多Taylor展开项以拟合共价键的伸缩势函数。例如,在MM2*共中**价键伸缩势函数在平方项的基础上增加了一个三次方项,
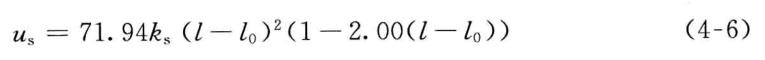
三次方项的引入虽能更好地近似偏离参考位置不大时共价键的伸缩势,但是,当分子初始构型远远偏离参考位置时,包含三次方项的共价键伸缩势可能取负值,或将导致结构优化等的失败。另外,只近似到三次方项的共价键伸缩势,拟合与共价键振动光谱对应的势能对键长的二次导数的效果不佳,经常显著偏离实验值。为了较好地解决上述两个问题,在MM3和MM4*共中**价键伸缩势被展开到四次项,
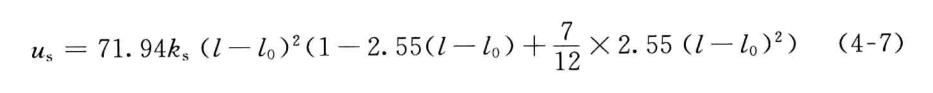
2.键角弯曲势
在MM2中,键角弯曲势也用两项表示。所不同的是,键角弯曲势除平方项外的另一项是六次方项,
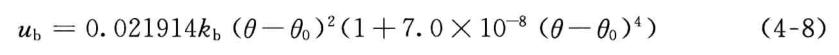
由于大多数有机化合物的键角偏离“参考值”较小,即使只保留平方项所引起的偏差也不显著。但是,对三元环、四元环等严重扭曲的分子,这样的误差就显得十分严重。因此,在MM3中键角弯曲能有更多的展开项,
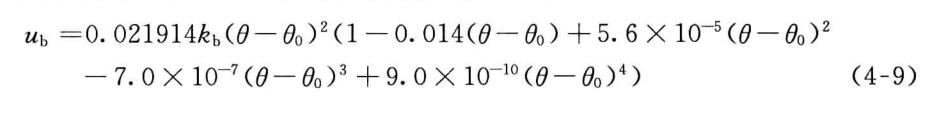
3. 二面角扭曲势
在MM1中,二面角扭曲势只有一项,但在MM2中二面角扭曲势包含三项,有关Vt.i和V..2的重要性可参考有关文献

4. 离面弯曲势与贋扭曲势
在MM3及更早的版本中,利用离面弯曲势来约束具有平面构型的结构单元对平面的偏离。对1—2—3—4四个原子形成的以原子2为中心的平面结构,1、3,4三个原子中的任意一个原子离开其余三个原子形成的平面,并形成一个角度X时,都将引起分子势能的升高。这样的离面弯曲势总共有三项,分别与离开平面的一个键对应,
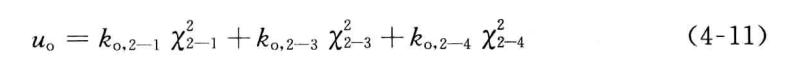
在MM4中,离面弯曲势被贋扭曲势所取代。贋扭曲势也包括三项,分别与三个贋扭角2—4—3—1、2—1—4—3、2—3—1—4对应,
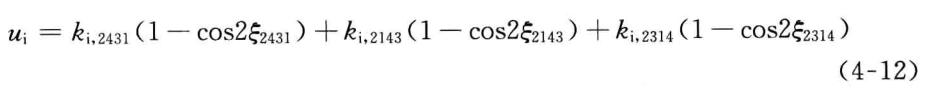
5. 伸缩-伸缩耦合势
共享一个中心原子的两个共价键Zl和 i2 之间的相互耦合势能为
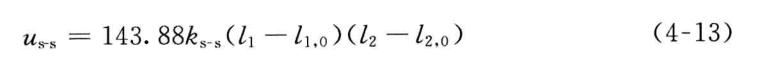
6. 伸缩-弯曲耦合势
伸缩-弯曲耦合是最重要的交叉项之一,也是MM2以及以后的版本中均引入的交叉项。以CH3—CH2—CH3为例,当中间碳上的两个C—H键收缩时.两个氢原子将靠得更近,使分子的能量有很大的升高。实际分子中,当两个氢原子向碳原子靠近时,键角H—C—H将略微增大,以消除这种影响。在分子力场中,这样的效应通过键伸缩-键角弯曲之间的相互耦合项来实现。在MMn中,这样的耦合项被写成
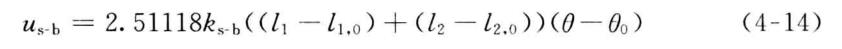
式中,A与仏分别为形成键角的两个共价键的键长;几和必为参考键长;0为键角;&为参考键角;怂b为耦合系数。
7. 伸缩-扭曲耦合势
虽然对大多数分子来说,伸缩-扭曲耦合并不重要,但对降茨烷等特殊的分子,该交叉项却对正确预报分子的结构非常重要。因此,MM3中引入了伸缩-扭曲势,事实上,即使在引入伸缩-扭曲势后,MM3对降茨烷结构的预报仍然不够理想。除此之外,在环丁烷中还引入了键角弯曲二面角扭曲之间的耦合丽,但在MM3中不再包括此项。

8.弯曲-弯曲耦合势
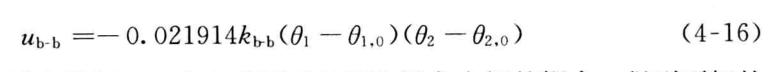
9.扭曲-扭曲耦合势

10.扭曲-贋扭曲耦合势

11. 弯曲-扭曲-弯曲耦合势

12. 贋扭曲-扭曲-贋扭曲耦合势

13.vanderWaals相互作用势
虽然Lennard-Jones12-6势函数被广泛应用于vanderWaals相互作用的拟合,但Lennard-Jones12-6势函数的排斥部分太硬,对精确拟合势函数不利。在MMn力场中,Allinger等使用Hill势函数拟合vanderWaals相互作用,
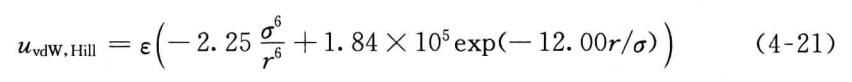
式中, a 为两个相互作用的原子的vanderWaals半径之和;e为vanderWaals相互作用的能量参数。在MM2中,指数项前面的系数为2.9X105,指数中的系数为12.5。值得注意的是,计算氢原子与其他原子间距离时,从碳原子外0.915A处计算,而不是氢原子核的位置。
14.静电相互作用势
在大多数分子力场中,静电相互作用以位于原子核位置的点电荷之间的库仑相互作用描述。但是,MMn力场中的静电相互作用则是以位于化学键上的偶极矩表示。Allinger等认为,以点电荷形式描述的静电势与以偶极矩形式描述的静电势没有本质区别。但对于蛋白质等非电中性分子(离子),MMn力场中包括了偶极矩-偶极矩相互作用、点电荷-点电荷相互作用、点电荷-偶极矩相互作用等。
MMn力场中,大多数分子中的C—H键、C—C键的偶极矩取0。但是,H—C(sp2)的偶极矩取0.6,C(sp3)—C(sp2)取0.95D(MM3中取0.90D)。
4.3.3MM4 的预报精度
实验测量或理论计算键长和键角的常用方法包括电子散射、微波谱测定转动惯量、量子化学第一性原理计算等。通过精心优化力场参数,拟合这些实验或理论数据,MM4对由重原子组成的共价键键长的预报精度可达〜0.004A,键角达l°o但是,由于与H原子有关的键长和键角的实验数据精度较差,MM4对相关键长和键角的预报精度也较差,一般对键长的预报精度在±0.5%左右。
