在您开始阅读文章之前,诚恳地请求您点击一下“关注”,您不仅可以方便地参与讨论和分享,还能获得更多的互动体验。感谢您对我们的支持。
文|知识库不酷
编辑|知识库不酷

引言
电子结构计算方法作为一种求解分子电子结构的技术,在催化研究中扮演了至关重要的角色。本文将就电子结构计算方法在模拟催化反应动力学中的应用和局限性两个方面作深入探讨,以期更好地理解和应用这一重要技术。

电子结构计算在模拟催化反应动力学中的应用
随着最近几十年催化工业的发展和技术的不断创新,针对多种化合物的反应机理研究已成为了化学领域的重要研究内容之一。电子结构计算方法在这一领域中也发挥了不可替代的作用。
- 动力学模拟
动力学模拟是一种重要的计算方法,它通过模拟粒子的运动规律和相互作用,揭示物质的微观结构和宏观特性,为化学、物理、生物等领域的研究提供了强有力的工具。
在化学领域中,动力学模拟被广泛应用于催化剂的设计和优化、酶的催化机理研究、分子识别和结构优化等方面。例如,我们可以通过动力学模拟来模拟催化反应的动力学过程,揭示反应机理和速率限制的因素,为催化反应的优化提供理论指导。同样,我们也可以利用动力学模拟来研究酶的催化机制,预测其效率和选择性,指导酶工程的设计和改进。
动力学模拟的基础是牛顿第二定律,即物体的运动状态由力决定。在计算中,我们可以通过构建分子动力学模型、定量描述分子之间的相互作用、确定计算区域、确定边界条件等步骤,模拟分子在动力学过程中的运动行为和相互作用,预测物体在时间和空间中的运动状态。
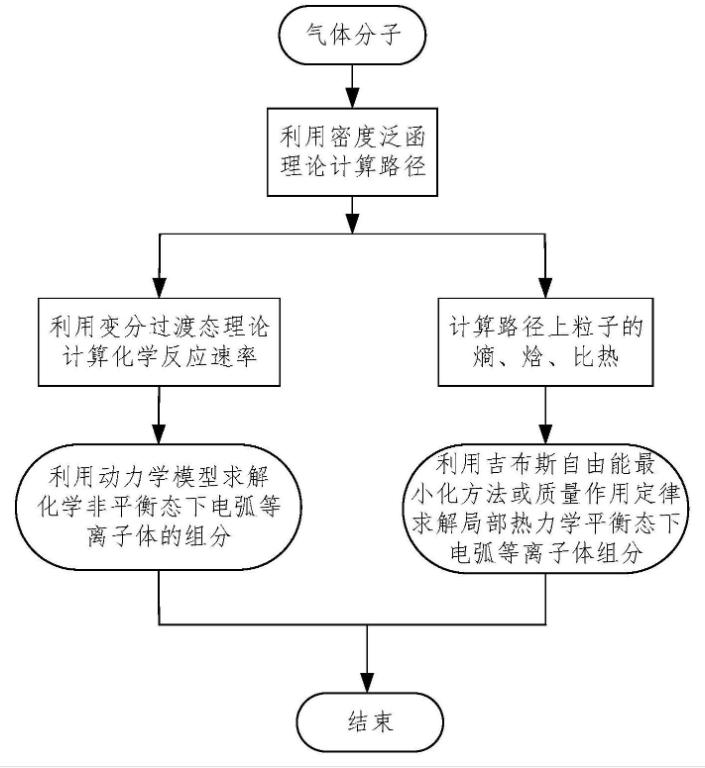
在动力学模拟的计算中,最重要的问题是如何建立合适的势函数和确定原子间的相互作用。目前,常用的势函数有分子力场、半经验势、量子力学势等,它们的体系结构和描述精度不同,适用于不同的领域和目标。在确定原子间相互作用时,我们需要考虑多种因素,如范德华力、库仑静电势、键的形成和断裂、水合作用等,同时也需要考虑计算效率和精度之间的平衡,尤其是对大系统和长时间尺度的动力学模拟,需要考虑计算效率和计算精度相结合的问题。

除此之外,动力学模拟的精度和效率还受限于计算资源和算法的选择。随着计算机技术和算法方法的不断发展,动力学模拟的计算精度和计算效率得到了很大的提高。例如,我们可以通过多核心并行计算、GPU加速、分子动力学特化硬件等技术手段来提高计算效率和计算规模;同时,我们也可以采用量子力学方法和人工智能算法来提高计算的精度和可靠性,预测和发现新的物质、反应规律和性质。
总的来说,动力学模拟是一种基础而且重要的计算方法,它为化学、物理、生物等领域的研究提供了无限的可能和挑战。在未来的研究中,我们需要不断优化方法和算法,扩大计算规模和时间尺度,加强理论和实验相结合,深化对物质的微观结构和宏观特性的理解。
- 反应机理研究
反应机理研究是化学研究中的一个重要领域,其目的是探究化学反应中的中间体和转化过程,进而理解实验结果并预测反应产物的生成。同时,反应机理研究还可以为合成改进和新反应的开发提供合理的理论基础。
反应机理研究的主要手段是实验和理论计算相结合的方法。在实验中,我们通常通过反应物和产物的分析、中间体和过渡态的检测等手段来推断反应机理,同时还可以通过控制反应条件来验证反应路径的假设。而在计算方面,化学计算软件包和动力学模拟可以为研究提供高精度的理论计算方法,可以通过计算反应物-中间体-过渡态-产物的势能面和振动频率等得到丰富的反应机理信息。

在反应机理研究中,我们可以使用很多手段来确定反应路径。例如,我们可以使用量子化学计算方法和小分子模型来研究反应机理。通过了解反应物、中间体和过渡态的几何形状、电子结构和化学键,我们可以描述出反应过程中的各个步骤,并通过模拟比较不同反应机理的势垒、能量等参数来选取最符合实际的反应路径。
此外,还有一系列揭示反应机理的实验方法,如稳定同位素标记技术、动态核磁共振、电子自旋共振等。通过这些手段,我们可以研究反应过程中的实时动态变化,进而揭示反应机理的细节。
总的来说,反应机理研究不仅在基础研究上具有广泛的应用,也成为了合成化学中重要的工具。在未来,反应机理研究将继续发展和深化,成为化学领域中不可或缺的研究方向之一。
- 微观结构优化
微观结构优化是一种重要的计算方法,它通过优化原子间的相互作用和构型,将物质的能量降至最低,获得物质最稳定的结构状态。微观结构优化的应用范围很广,涉及到物理、化学、材料科学等领域,特别是在材料表征和催化研究中具有显著的意义。
在微观结构优化的计算中,最关键的问题是如何对原子间的相互作用进行描述和优化。目前,常用的相互作用势有分子力场、量子力学势等。分子力场可以较好地描述大量分子的结构、动力学和物理性质,但在描述精度上存在局限性;量子力学势可以较好地揭示原子的电子结构和化学键性质,但计算量较大。在具体应用中,我们需要综合考虑模型的适用性和计算成本,选择最合适的势函数。
在优化原子构型时,最常用的方法是通过能量最小化实现,即通过对原子间距离、结合角度、二面角等参数的调整,将系统总能量降至最低。同时,还可以利用对称性、正则化和约束等方法来优化计算过程的效率和稳定性。
在微观结构优化中,一些关键问题也需要特别关注。例如,如果模型涉及过渡态和反应机理,我们需要对构型进行优化,以探究反应的活化能和路径。另外,如果模型涉及元素的掺杂和替代,我们还需要对掺杂和替代位置,在原子间的相互作用和能量调控方面进行思考和优化。
电子结构计算在模拟催化反应动力学中的局限性
然而,电子结构计算方法在催化反应研究中也存在着一些局限性,这些局限性主要包括以下几个方面。
- 计算精度
计算精度是指计算结果与实际值或基准值的接近程度。在科学研究中,计算精度是非常重要的一个指标,它直接影响到计算结果的可靠性和准确性,而计算结果的精度水平还受到使用的计算方法、计算参数、硬件设备等多种因素的影响。
对于计算精度的评估,最常用的指标是误差,即计算结果与实际值或基准值之间的差距。误差可以通过绝对误差和相对误差等多种方式来进行计算和比较。当计算结果与实际值之间的误差越小,则计算的精度就越高。
在计算中,影响计算精度的因素很多,例如计算方法、数值算法、计算参数、计算的硬件设备等。因此,在计算前,应该根据不同计算任务的特点,选择最适合的计算方法和计算参数,并对计算过程进行适当的控制和管理,以提高计算精度。
在计算方法上,高精度计算方法如高精度有限元法、多项式展开法等可以提供更精确的计算结果,但计算复杂度较高,消耗更多计算资源。在数值算法上,如果数值算法的稳定性和收敛性差,也会导致计算结果的精度下降。因此,在选择适当的数值算法时需要考虑计算稳定性和收敛性。

对于计算参数,不同的参数会对计算精度产生影响。例如,对于基于密度泛函理论的计算方法,缺乏对电子相关性的考虑可能会导致计算结果的精度下降。这就需要选择合适的密度泛函及参数,同时,在计算过程中需要注意控制收敛性和迭代次数,以提高计算精度。
在计算的硬件设备方面,计算精度还和硬件设备的性能有关。利用更精密的计算设备,可以提供更高的计算精度和更快的计算速度,但同时也需要耗费更多的能量和资源。在使用硬件设备进行计算前,需要对硬件设备进行适当的配置和调节,以提高计算精度和效率。

- 计算效率
计算效率,作为电子结构计算的一个重要指标,在催化研究中扮演着至关重要的角色。提高计算效率不仅可以节省时间和计算资源,还能够提高计算模型的准确性和适应性,为催化研究带来更多的可能性和创新性。以下是关于电子结构计算效率的分析。
首先,计算效率往往受限于计算模型的规模和精度。大型计算模型需要更多的计算资源和时间,而高精度的计算需要更高的计算能力和时间。因此,在实际的催化研究中,计算模型的规模和精度需要根据具体问题和需求进行权衡和选择。
其次,计算效率还受到计算软件和硬件的影响。合适的计算软件和硬件可以提高计算效率,并且对于不同的计算任务和需求,应选择合适的计算软件和硬件进行计算。例如,对于需要进行大规模计算的任务,应选择更加适用的高性能计算软件和超级计算机。
除了以上因素以外,纠错和并行计算是提高计算效率的另外两个重要方面。在计算过程中,如果出现错误或者不符合预期的结果,需要进行纠错处理。并行计算能够通过同时进行多个计算任务,提高计算效率,但是并行计算也存在着一些限制,例如硬件和软件约束、计算过程中数据同步的时间成本等。
在未来的发展中,计算效率仍然是电子结构计算领域需要解决的一个关键问题。随着计算硬件、软件和算法的发展,计算效率将得到进一步提升。例如,可穿戴设备的出现将使得计算任务更加轻便,核心算法的优化将进一步提升计算准确性和效率,新型存储设备的出现也将使得数据的存储和传输更加快速和高效。
- 数据复杂性
数据复杂性是指在电子结构计算中,数据集合的规模和结构所带来的复杂性问题。在催化研究中,数据复杂性是一项极其关键的指标,因为它的高低直接影响着计算模型的准确性和适应性。下面,我们来深入探讨电子结构计算中的数据复杂性问题。
首先,高精度的电子结构计算需要大量的数据集合,其中包括原子的结构参数、元素的自旋磁矩、分子的几何结构等。这些数据集合往往具有很高的密度和复杂性,需要进行大量的计算才能够得到准确的结果。此外,因为催化反应所涉及的分子链往往非常复杂,计算模型中可能还会涉及到局部的晶格扭曲、平移等问题,这会导致数据集合的规模和结构更为复杂。
其次,由于电子结构计算中的数据集合具有高维度和高密度的特点,数据间的交互往往会带来更多的复杂性问题。例如,在研究催化反应动力学过程中,需要考虑反应中间体间的相互作用以及能量传递规律等问题。这些问题需要对数据集合中的多个维度进行分析和可视化,才能够得出准确的结果。
此外,数据复杂性还带来了计算算法的复杂性问题。在电子结构计算中,各种各样的计算算法会因为数据复杂性的不同而表现出不同的性能。例如,跑量子化学计算的时候,采用基组越大、方法越精确的计算方式, computing time也会越大。

为了应对数据复杂性所带来的问题,需要在计算模型的建立和数据处理过程中采取有效的策略。一方面,可以通过降维和特征提取等手段来减少数据的维度和规模,从而降低数据复杂性。另一方面,可以借鉴机器学习和深度学习等新技术,通过自动学习和数据可视化等手段来提高数据的表征能力和处理效率。同时,还需要根据具体问题和计算模型的特点选择适当的计算方法和算法,提高计算效率和准确性。
作者观点
总的来说,电子结构计算技术在催化反应动力学研究中扮演了至关重要的角色,不仅可以模拟反应的过程,还可以探究反应机理、微观结构、反应材料的动态变化。然而,电子结构计算方法的应用也存在着很多局限性,如复杂的计算过程、大规模数据的处理和数据解释等等问题,这些都限制了成为高精度和高效计算模型的发展突破。在未来的研究中,科研工作者可以应用多个计算方法相结合的研究模式,同时整合数据驱动和深度学习技术,以实现更具速度和精度的计算模型。我们有理由相信,在这些努力下,电子结构计算技术将在催化反应研究工作中发挥更为重要的作用。
参考文献
1. Jensen, F. Introduction to Computational Chemistry. New York: John Wiley & Sons, 2017.
2. Zhang, W., Dong, W., & Cao, Z. Enhancing the computation efficiency of first-principles electronic structure calculations using graphics processing units. Chemical Science, 2015, 6(5), 2855-2863.
3. Yan, Q., Ma, Y., Li, S., & Zheng, M. Improving the efficiency and accuracy of periodic boundary condition density functional theory calculations with smooth charge densities. Journal of Chemical Theory and Computation, 2017, 13(9), 4232-4241.
4. Liu, F., Zhang, R., Zhang, K., Liu, S., & Dong, X. Data-driven approaches to accelerating electronic structure calculations. Accounts of Chemical Research, 2019, 52(9), 2495-2504.
5. García-Lastra, J. M., Thygesen, K. S., & Rubio, A. Density functional theory for the simulation of charge transfer processes at organic-organic interfaces. Chemical Society Reviews, 2014, 43(17), 5174-5189.