第一部分 分子结构和热力学
第一章 导论分子结构和化学键模型
国外的大部头教材为了使化学看起来像是门理科,大都会从量子力学讲起,这本也不例外,其实波粒二象性、薛定谔方程解这些东西吧,化学向教材里表述的都一样,反而是早年间的《福井谦一有机量子化学》这些有点自己的理解。现在还有很多有机化学书会讲价键模型,所以本书也花了些篇幅介绍了,1.2之后才会讲一些现代概念,(这篇笔记讲不到,至少在下一篇中才会讲到),因为前面的传统内容已经讲烂了。现在的高中*党**化竞*党**绝对比我懂,限于本篇笔记的篇幅,就不详述了。
1.1 化学键的基本概念
1.1.1 量子数和原子轨道
Bohr模型、主量子数、角量子数、自旋量子数,s轨道和p规定、自旋配对问题
提到了“ 电子相关效应 是指一个电子可以感应另一个电子的运动轨迹从而改变它本身的运行路线,这种能力使库仑排斥作用最小并保持系统的能量最低。”这应该是为了和后面的超共轭效应呼应。
1.1.2 电子构型和电子结构图
基态、激发态、能量最低原理、Pauli原理、洪特规则、简并轨道=拥有相同能量的轨道
1.1.3 Lewis结构
1.1.4 形式电荷

对有机化学而言记住几个经典结构就可以了,形式电荷只是一种记录方法,有机化学中其实不算实用。

1.1.5 价电子对互斥规则
这个原理来自简化的静电排斥模型,是一个基础模型,大部分时候尤其是对于有机物而言都是对的。另外注意当中心原子为过渡元素时(具有全满、半满或全空的 d轨道的除外),VSEPR理论不准确。
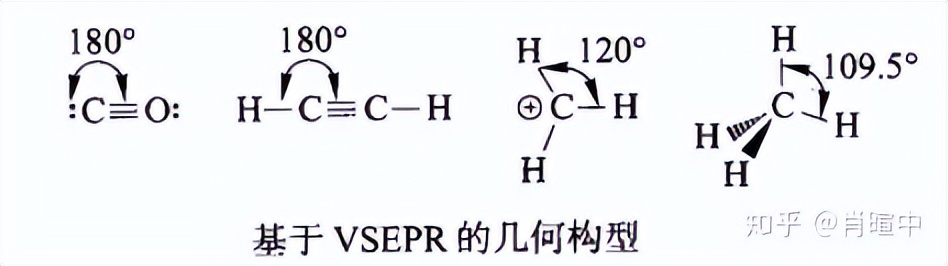
由于取代的不同,实际测量的数据和以上结构之间会有偏差,一个经典的例子就是甲烷-氨-水键角的比较,但注意电子对“大小”这个概念只是个合情合理的推论,孤对电子的尺寸是不是比成键电子+链接的基团大,这一问题目前还有争议。
作用:预测几何构型,立体效应,立体位阻。
1.1.6 杂化
Pauling的杂化理论和VSEPR是两个东西,虽然经常联合使用或相互比较,但出发点完全不同。完全理解杂化需要一些结构化学的知识,但现在高中化学已经引入了这个概念,线性组合有讲不清,有时只好搬出VSEPR。另外解薛定谔方程不会直接得到Px,Py轨道。

杂化指数 的概念经常出现在讨论弯曲键,亲核/亲电中心软硬程度等问题上,很多时候看到 sp3.4 之类的概念会感觉摸不着头脑,其实它的逻辑是:从sp杂化到sp³杂化,s的成分越少,键角越小,所以这个概念是用来补充上面提到过的VSEPR模型偏差的。很多时候是由观察结果逆推的,现在因为杂化概念渐渐弱势,这个说法也没什么重要的了,更多是用在教学中。不过现在因为有时会和NBO(自然键轨道)的结果对照,而且用得还是不少,知道一些也好。

二维核磁谱和杂化指数的关系。“s轨道在碳原子核周围电子密度高”、“p轨道更适合于形成键角小的键”,很多地方会把上面这个结论推广到推拉电子问题上。
1.1.7 化学键的价键/分子轨道混合模型
有机中化学键理论主要涉及 价键理论VBT 和 分子轨道理论MOT ,详细讲这两个太花时间,可以翻翻结构化学或国外的物理化学教材,注意”Lewis结构式-电子局域化-共振论-杂化论-电负性/电子亲合势-杂化“这一整套是Pauling的理论体系,MOT则是另一套,现在为了更形象地解释一些问题会把两者结合在一起,两者也确实相辅相成,但用MOT讨论共振式其实很奇怪。
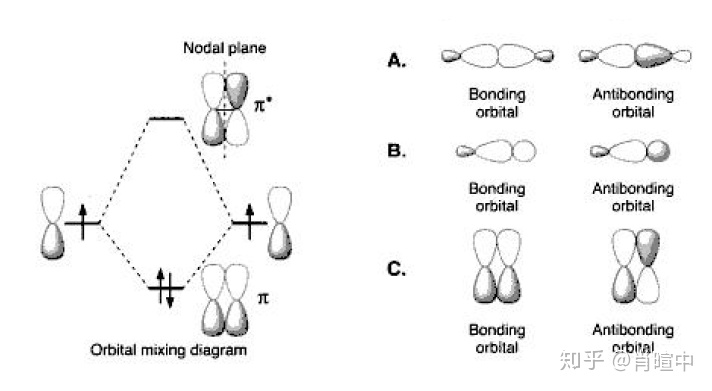
高中讨论的σ键和π键属于VBT理论,而成键轨道反键轨道属于MOT,不过现在两者也确实常常联用,因为分子轨道线性组合的计算对大多数人还是太抽象了,‘结合’VBT的杂货轨道可以方便理解。Nodal plane-节面,Bonding orbital-成键轨道,antibonding orbital反键轨道。左图表示的是乙烯中的π键。
σ 键和π 键的形成包括位于相邻原子之间的成键和反键分子轨道,这已经是有机化学家最常用的标准概念。大多数有机化合物的反应特性可以用这种概念很好地解释,它是描述有机反应机理的电子推动方法的出发点 。这种化学键理论在有机化学中极其重要。
这张Evans讲义的截图就是上面这段话的一个例子。这里讨论的是卤代烃E2消除的立体化学问题,电子推动的方向是β碳上的HOMO轨道→α碳上C-X键的LUMO轨道,后面应该会细讲这种东西。
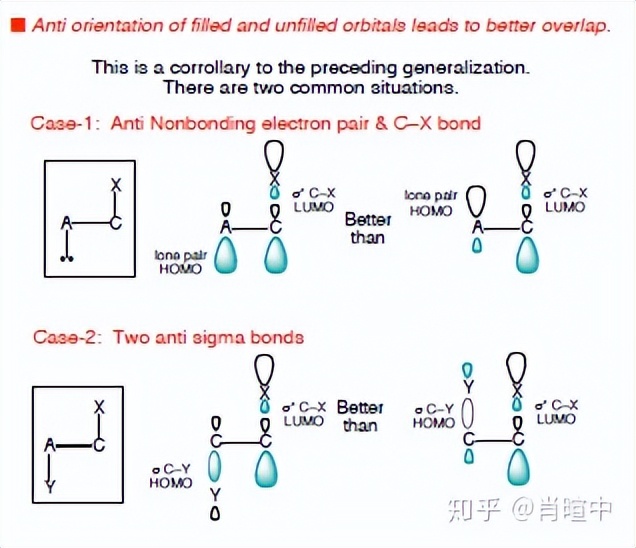
VBT和MOT都是以计算为出发点理论,以建立模型-实验证实为主,它们具有很高的理论指导性,所以没有之前的理论种那种根据实验结构修修补补的感觉。
1.1.8 极性共价键。
预测化合物的反应特性需要知道化合物的电荷分布。前面讲了VSEPR 和杂化等概念可以用来解释分子的形状和结构,现在利用这两个理论推测分子内的电荷分布,这是非常经验化的东西,也正是因为有这样的东西,给人一种有机靠背诵的感觉。虽然现在有计算化学软件,比如高斯软件,可以给出分子的HOMO-LUMO,给出各种可能机理的中间态能量,但是每看到分子反应都用高斯算太费事了,希望随着AI的发展以后有个程序能自动给出这个分子的反应特性。怎么说是因为很多反应的结果确实反直觉,只能看结果推测原因,比如青蒿素还原过程成二氢青蒿素的过程中过氧键可以保留。
几个概念 共价键 (极性共价键)、 电负性 (Pauling电负性,离子势,电子亲和势)不详述,这里提一点,Fr的金属性不如Cs,理由和6s惰性电子对效应一样。
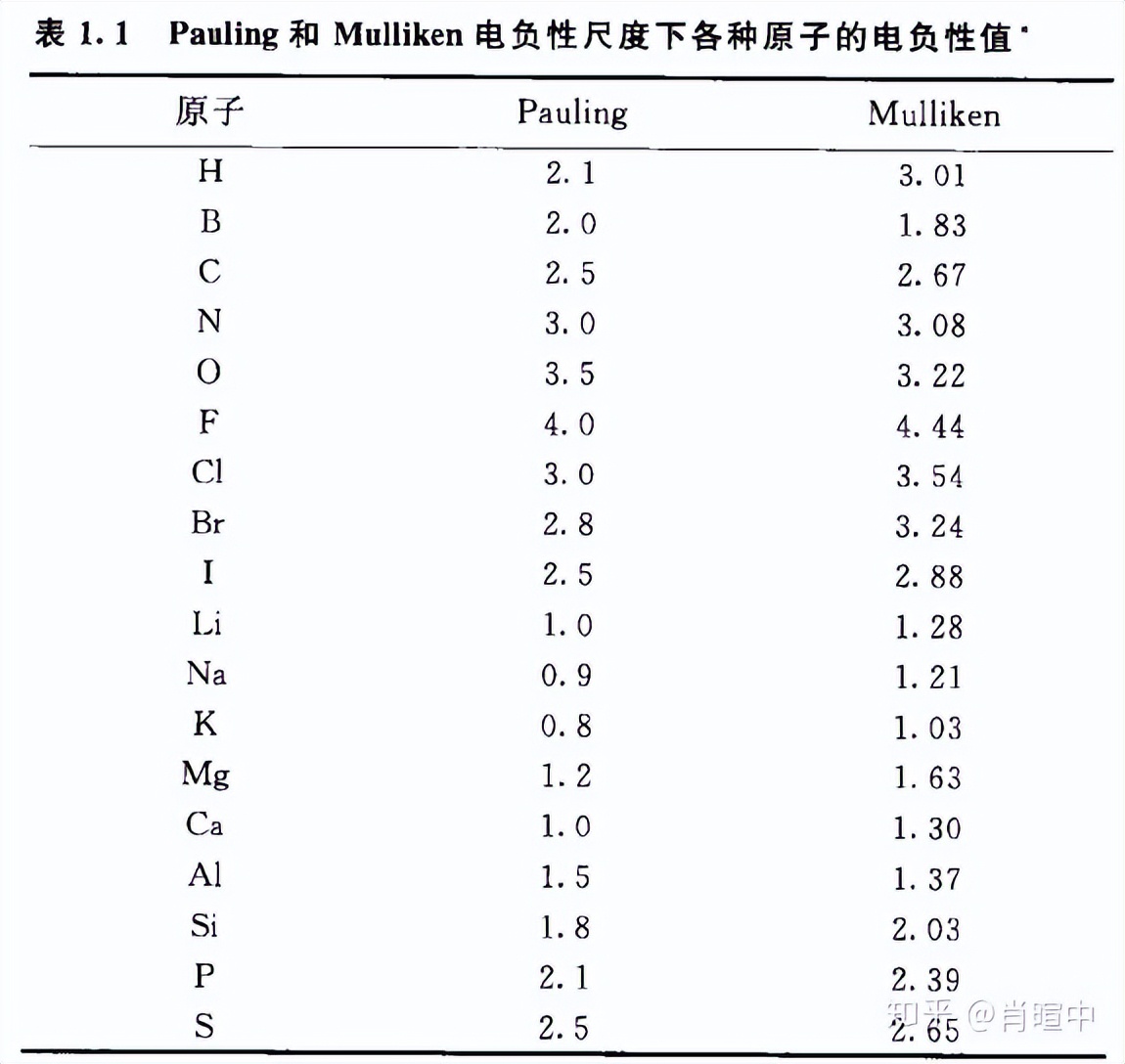

2019最新的电负性表J. Am. Chem. Soc. 2019, 141, 342−351
部分电荷: 当一个比碳原子电负性更强的原子与碳原子成键时, 键上的电子将偏向电负性更强的原子 ,造成在这个原子上带部分负电荷,而碳原子上带部分正电荷。相反,当该原子的电负性比碳原子更弱,碳原子将拥有部分的负电荷。这些部分电荷用δ+ 和δ- 表示:

注意 部分电荷 和 形式电荷 是两个概念,电负性差别为1. 7 时,可以认为是50% 的离子键和50% 的共价键。而当电负性差别大于1. 7 时,可以认为是离子键。按照这种观点, Li ,Na , K 与F ,Cl, Br , 1 形成的键为离子键,但一般不强调这个。
由于Pauling的电负性表流传很广,出现了很多奇怪的结论,比如C-I键极化程度比C-H键小,S-O、P-O键极化程度比C-O小,后来更新的电负性表可以解释以上问题。
静电势能面
本节开头讲了,电负性-部分法则属于”半经验规则“,遇到复杂体系将难以处理,比如乙酸乙酯中的两个氧是δ-,氧相连的碳是δ+,这样复杂的部分电荷对推测化学反应没有帮助。
静电势能面能够更加深入地展示分子内的电荷分布,它们来自于对分子电子结构的量子力学计算,完全没有使用电负性、杂化、键偶极或者任何共价化学键模型的描述性特征,不依赖于经验观察的量子力学计算,依据它们的结果可以估计分子内的电荷分布。
在这种图首先会给分子一个表面。该表面与van der Waals 表面相似是通过把每个原子当成一个半径等于其van der Waals 半径的球绘制出来的。静电势能面是一个等密度面,即表面上电子密度处处相等,比如0.002 个电子/Ų。一般来说,负的静电势标红色,正的静电势标蓝色。
”静电势“的概念和物理上一样,可以想象用一个带+1 电荷的非常小的球在等密度面上滚动,通过判断小球是被表面吸引还是被排斥,以及受到作用的能量大小是多少,给出静电势的数值。看静电势能面的图时一定要注意尺度问题。
但注意基态静电势能面只能帮助我们理解分子电荷分布,预测反应过程还是需要计算激发态/过渡态的静电势能面。因为当一个化学反应发生时,一般会发生显著的电荷重新分配。例如,当一个负离子亲核试剂加成到*酮丙**的醛基上时,过渡态的静电势能面与基态下*酮丙**分子的明显不同。因为反应过程是由过渡态来决定的,直接使用这些基态的静电势能面来预测或者解释反应是有风险的。
诱导效应
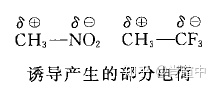
一般有机书上都有,由于电负性不同的取代基(原子或原子团)的影响,使整个分子中的成键电子云密度向某一方向偏移,使分子发生极化的效应。这个效应经常和 场效应 相比较,但很多时候都是受两者共同作用。下面这个例子很经典,但这个例子也让一些人把诱导效应和共轭效应搞混了。

基团电负性 没什么好说的

杂化效应
之前说的杂化指数问题就在讲s轨道电子和p轨道电子的区别,一般认为:
电负性:sp>sp²>sp³
一个典型的例子就是乙炔、乙烯、乙烷氢的pKa问题,书上的例子是氟甲烷:
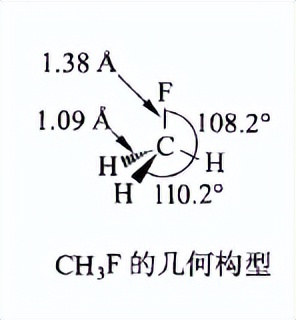
H-C-F 键角的减小是由于F 是最强的电负性取代基。F 倾向于与包含更多p 轨道成分的C 原子杂化轨道成键,因为从C 原子的p 轨道吸引电子比从C 原子的s 轨道吸引电子更容易。通常所说的s 轨道比p 轨道具有更好的电子穿透性,表示从s 轨道吸引电子更加困难。
1.1.9 键偶极、分子偶极和四极矩
键偶极、偶极矩的定义和单位、偶极矩的方向、偶极相反端相互吸引 不详述,注意键偶极在判断分子反应机理时很重要,结合部分电荷概念可以推测反应过程。这是一个可测的概念,在溶剂化问题和分子识别问题中也有应用。
分子偶极矩
分子固有属性,可测,是键偶极的 向量和。
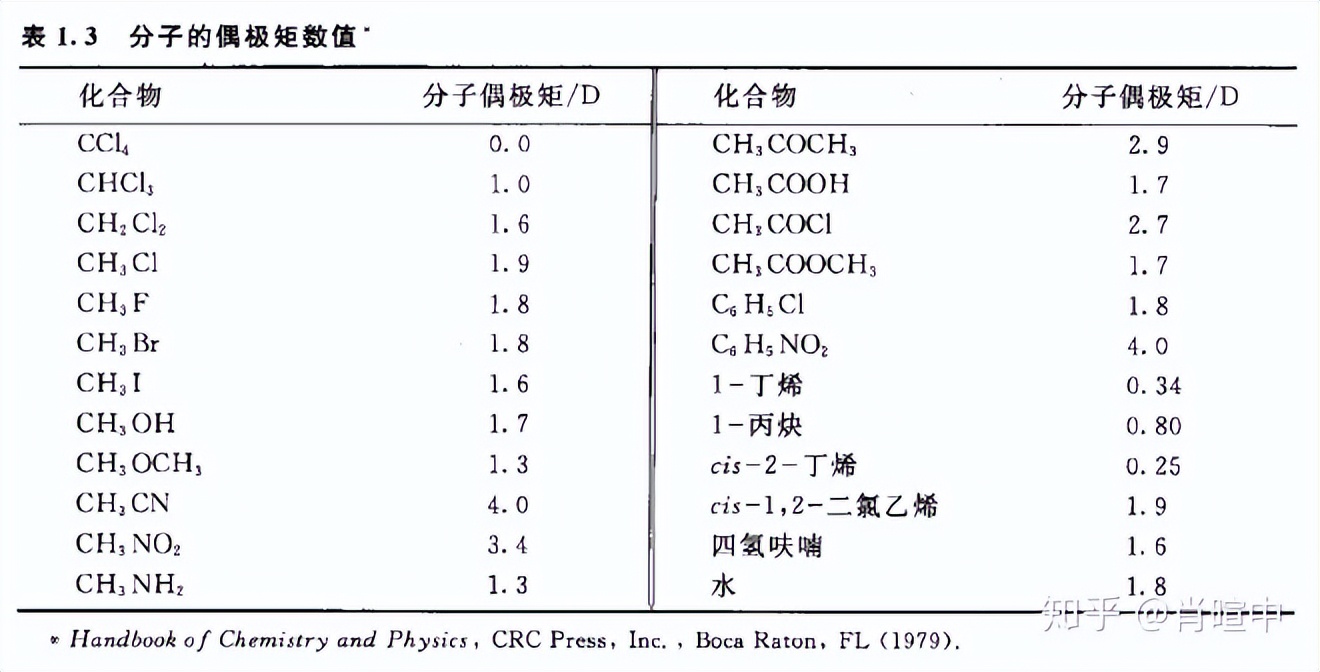
最经典的问题就是一氯甲烷,二氯甲烷,氯仿,四氯化碳的偶极矩比较。

分子四极矩 也是测试出来的东西,在光谱学中比较重要,有机反应中涉及不多,但还是要贴出来,这部分资料不常见。
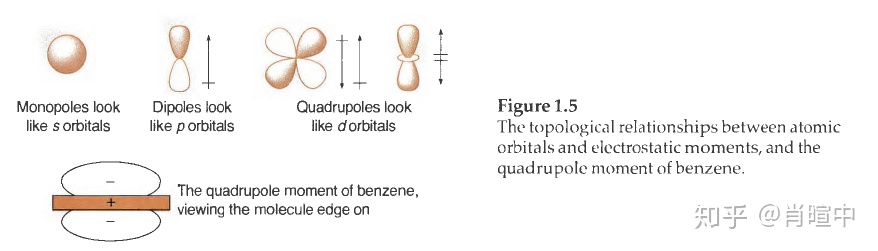
学过拉曼光谱和核磁光盘的话应该知道,在对分子电荷分布的完整叙述中,偶极只是级数展开中的一项,应该包括:单极、偶极、四极、八极、十六极……
单极只是一个点电荷,对于离子而言该项占主导。一般中性分子,只讨论偶极,后面的可以忽略,但有时四极也很重要。
四极 是两个偶极的特定排列,使得没有净偶极存在。如果有净偶极存在,就称为偶极,而不是四极。有趣的是,多极的展开遵循一种熟悉的模式:单极看起来像s 轨道(球形) ,偶极看起来像p 轨道( 一个正端和一个负端) ,四极看起来像d 轨道,八极看起来像f轨道,等等。但是静电矩和轨道之间的类比仅仅是为了解释相位性质。轨道并没有极性特征。
一个简单但不一定准确的说法是
(1)偶极矩:1维空间中-q和+q两个点电荷,两者的中点位于原点。
(2)四极矩:2维空间中两个-q和两个+q,共4个点电荷交替地占据正方形的4个角,正方形的中心在原点。
(3)八极矩:3维空间中4个-q和4个+q,共8个点电荷分别交替地占据立方体的8个角,立方体的中心在原点。
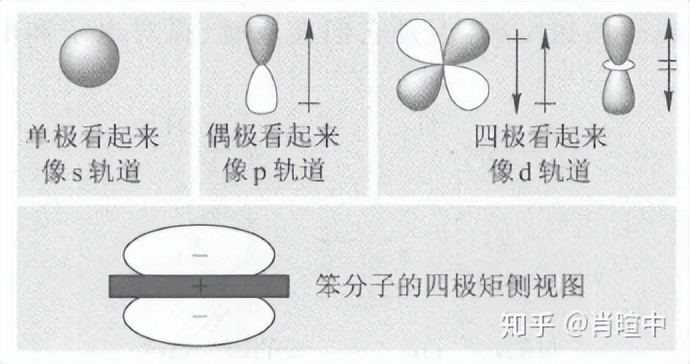
写成”笨“也是服了
有机化学中最常见和最重要的四极矩是苯分子中存在的四极矩。实验测定苯分子拥有一个很显著的四极矩,其电荷分布如图1. 5 所示,也可以看前面的静电势能面图。就像分子偶极矩那样,我们能够将分子的四极矩解释为键的偶极之和。在这个例子中,将六个C(δ-)—H(δ+) 偶极相加得到分子的四极矩。苯分子中存在显著的永久性四极矩是sp² 杂化的C 比H 电负性更强的确凿证据。必须用六个C(δ-)—H(δ+)偶极来解释该现象。注意,环己烷的四极矩几乎可以忽略不计,表明sp³ 杂化的C 和H 的电负性相似。