
一、法规中对于病例报告表的定义
1.《医疗器械临床试验质量管理规范》(2022年第28号):
病例报告表,是指按照医疗器械临床试验方案所规定设计的文件,用以记录试验过程中获得的每个受试者的全部信息和数据。
2. ICH-GCP:

按试验方案所规定设计的一种印刷的、光学的或电子的文件,用来记录每一名受试者在研究过程中的全部信息报告给申办者。
二、如何设计一份良好的病例报告表?
1. 明悉病例报告表的两大目的:
(1)回答方案的基本假设(即有效性和安全性):要求设计者透彻理解方案,使得对于方案中各访视试验流程,CRF都能有相应数据表格作为记录载体,从而能完整准确地采集记录研究方案和CSR种所要求采集的数据。
(2)用于试验管理和记录法规、GCP依从性的支持性证据:要求CRF收集临床研究中相关法律法规对数据的要求,如AE/SAE。
2. 明确设计病例报告表的基本原则:
(1)依从药政监管要求
(2)理解和遵循试验方案
(3)了解临床研究报告的要求
(4)明确收集数据的指标定义
(5)做到全面完整、简明扼要
(6)避免主导性问题的出现
(7)便于使用者的填写和录入
3. 了解病例报告表的设计流程:
CDASH推荐的CRF设计流程
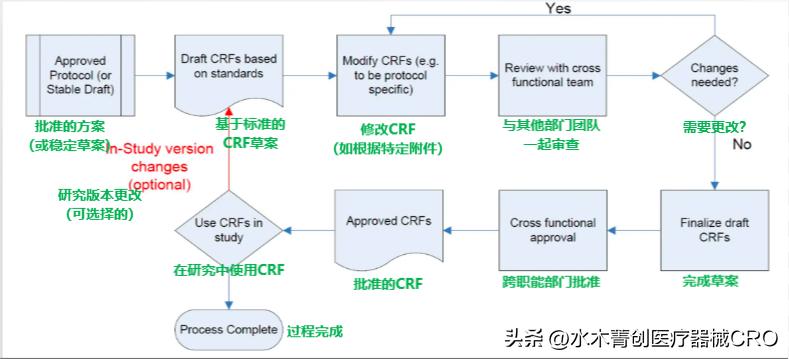
三、如何做好病例报告表的质量管理?
1. 建立良好的文件管理系统加强对病例报告表的质量保证
2. 进行临床试验项目各方多层次审阅
3. 完善CRF的审核和批准流程,并保存好批准记录
4. 各职能部门成员尽各自的职责
|
人员 |
主要职责 |
|
CRF设计者 |
确保数据收集的完整、正确、与方案的访视计划一致,以及CRF审阅及修改过程记录在CRF审核意见日志中 |
|
临床医生 |
确保疗效和安全性指标和变量的准确,通常会给CRF设计者/数据管理人员提供这些数据的类型和收集方法 |
|
监查员 |
确保数据清晰无误,易于研究者的填写 |
|
统计师 |
对照分析计划,确保收集的数据点满足统计分析需求 |
|
数据管理人员 |
从数据录入和清理的角度确保数据逻辑性和合理性的质量 |
|
数据建库员 |
数据收集的方式,变量类型不影响编程,确保系统及其数据库符合CDISC标准 |
|
药物安全警戒人员 |
安全性数据的收集方式有利于药物安全报告 |
|
质量保证员(QA) |
运用QA检查计划确保整体质量及其合规性;确保所有CRF文件记录存档备查 |
|
监管部门 |
确保CRF的设计符合GCP法规 |
5. eCRF质量管理的核查流程
(1)版面设计核查
(2)访视页面是否和数据库列表一致
(3)数据录入测试
(4)单变量核查
(5)报告的数据检测
(6)不在eCRF页面中显示的默认值检测

水木菁创医疗器械临床试验CRO
为中国医疗器械研、检、产、商提供专业支持
北京水木菁创医药科技有限公司是专注于为医疗器械企业提供专业临床研究技术服务的合同研究组织(CRO)。主要 为医疗器械(含体外诊断试剂)企业提供医疗器械临床试验、医学撰写、全球注册、GMP咨询等专业化技术服务与一站式解决方案 。
总部位于北京,分别在华南、西南、华东设立分公司,并在国内多个城市派驻人员,形成完善的、覆盖全国的服务网络。
团队精通于上市前后各类临床试验、注册及临床评价工作,参与过众多项目的执行与策划,实践经验丰富。团队成员过往经验累计服务客户超过3100家,累计取得注册证超过1080张。