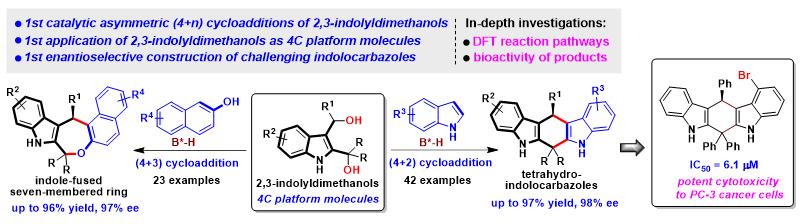
石枫团队代表性工作:吲哚二甲醇参与的催化不对称(4+n)环加成反应
本文来自微信公众号:X-MOLNews
导语:
手性吲哚并环骨架属于一类重要的杂环骨架,催化不对称构建该类手性骨架一直是合成化学领域的重要目标之一。吲哚甲醇是一类适用于催化不对称构建手性吲哚骨架的多功能平台分子,吲哚甲醇化学已经成为一个新兴的研究领域。但是,其参与的催化不对称环加成反应都是基于单吲哚甲醇的反应特性,而吲哚二甲醇作为一类新型的吲哚甲醇,其反应特性却鲜为人知,其参与的催化不对称环加成反应更是属于未知的非常具有挑战性的化学。为了解决这一挑战性问题, 江苏师范大学/常州大学石枫、谭伟 等人 提出了2,3-吲哚二甲醇作为四碳(4C)平台分子的概念 ,设计并实现了 首例2,3-吲哚二甲醇参与的催化不对称(4+2)和(4+3)环加成反应,高收率、高区域选择性和对映选择性地构建了手性吲哚并六元环和七元环骨架,并且发现了一些具有显著抗肿瘤活性的手性吲哚并环分子 。该工作不仅代表了吲哚二甲醇参与的第一例催化不对称环加成反应,实现了其作为4C平台分子在不对称催化合成中的应用,而且实现了具有挑战性的手性吲哚并咔唑和手性吲哚并七元环骨架的高对映选择性构建,为构建手性吲哚并环骨架提供了一种高效的策略。更重要的是,该工作对吲哚二甲醇参与反应的机理、中间体和活化模式进行了深入探究,为理解该类新型吲哚甲醇的反应特性提供了理论依据,促进吲哚甲醇化学从单吲哚甲醇朝着吲哚二甲醇这一更高、更广的方向深入发展。相关论文发表于 Angew. Chem. Int. Ed. 。
手性吲哚并环骨架属于一类重要的杂环骨架,广泛存在于天然产物和药物活性分子中。图1列举了一些含有手性吲哚并五元至七元环骨架的天然产物和生物活性分子。因此,催化不对称构建手性吲哚并环骨架一直是合成化学领域的重要目标之一,同时也是手性吲哚化学永恒的主题。

图1. 一些含有手性吲哚并环骨架的天然产物和生物活性分子
化学工作者们朝着这一目标付出了巨大的努力,发展了多种催化不对称构建手性吲哚并环骨架的方法。江苏师范大学/常州大学 石枫 团队致力于手性吲哚化学领域,针对该领域的科学问题,提出了“设计新型的吲哚平台分子及其参与的催化不对称反应”这一研究策略,实现了多种手性吲哚骨架的高效、高对映选择性构建(工作小结: Acc. Chem. Res . 2020 , 53 , 425; Acc. Chem. Res . 2022 , 55 , 2562)。其中,吲哚甲醇已被公认为是一类适用于催化不对称构建手性吲哚骨架的重要平台分子(综述: J. Org. Chem . 2017 , 82 , 7695; Adv. Synth. Catal . 2020 , 362 , 1214; Chin. J. Org. Chem . 2022 , 42 , 3351)。例如,化学工作者们已经发展了3-吲哚甲醇、2-吲哚甲醇、3-取代-2-吲哚甲醇以及其他吲哚甲醇,将其作为催化不对称环加成反应的多功能平台分子。事实上,吲哚甲醇参与的催化不对称环加成反应已被证明是对映选择性构建手性吲哚并环骨架的强有力方法(图2)。图2a归纳了代表性吲哚甲醇及其在环加成反应中的特性,通常而言,在手性布朗斯特酸(B*-H)存在下,这些吲哚甲醇可以脱水产生多种碳正离子和乙烯基亚胺正离子中间体,作为三原子合成砌块发生对映选择性(3+n)环加成反应。基于该反应特性,实现了一系列3-吲哚甲醇参与的催化不对称(3+n)环加成反应(eq.1)(代表性例子: Chem. Commun . 2014 , 50 , 15901; Chem. Eur. J. 2014 , 20 , 11382)。2-吲哚甲醇则显示出双重反应性质(C3-亲电性和亲核性),实现了具有不同区域选择性的催化不对称(3+2)、(3+3)和(4+3)环加成反应(eq.2)(代表性例子: Adv. Synth. Catal. 2016 , 358 , 3797; Angew. Chem. Int. Ed. 2019 , 58 , 8703; Angew. Chem., Int. Ed . 2021 , 60 , 2355)。此外,3-取代的2-吲哚甲醇作为碳-碳-氮(CCN)合成砌块也可以参与催化不对称(3+2)和(3+3)环加成反应(eq.3)(代表性例子: Org. Lett . 2016 , 18 , 5660; Adv. Synth. Catal . 2017 , 359 , 2660)。
虽然吲哚甲醇化学得到了迅速发展,但是已发展的催化不对称环加成反应都集中于单吲哚甲醇。与之形成鲜明对比的是,含有两个羟基的吲哚二甲醇的反应特性却鲜为人知,其参与的催化不对称环加成仍然属于一个未知的化学(图2b)。考虑到吲哚二甲醇的两个羟基可能会赋予这类平台分子一些新的反应特性,石枫团队设计了2,3-吲哚二甲醇作为一类新型的吲哚甲醇平台分子( Org. Chem. Font . 2018 , 5 , 2657),期望实现其在构建手性吲哚骨架方面的应用。从理论上而言,2,3-吲哚二甲醇可以作为1,4-双亲电试剂,在B*-H存在下与双亲核试剂(Nu1-Nu2)发生催化不对称(4+n)环加成反应,从而构建手性吲哚并环骨架。但是,在该类反应中存在一些挑战性问题,主要包括:(1)调节2,3-吲哚二甲醇中两个羟基的反应活性,以控制环加成反应的区域选择性;(2)寻找合适的双亲核试剂作为反应物,以实现催化不对称(4+n)环加成反应;(3)发现高效的手性B*-H作为催化剂,以控制反应的对映选择性;(4)揭示反应的途径和活化模式,探究产物的生物活性。

图2. 吲哚甲醇参与的催化不对称环加成反应的研究概况及存在的挑战
为了解决这些挑战性问题,石枫团队基于对吲哚甲醇化学的理解(代表性工作: Angew. Chem. Int. Ed . 2017 , 56 , 116; Angew. Chem. Int. Ed . 2019 , 58 , 8703; Chin. J. Chem . 2020 , 38 , 543; Angew. Chem. Int. Ed . 2021 , 60 , 2355; Chin. J. Chem . 2022 , 40 , 2151),提出了发展2,3-吲哚二甲醇作为四碳(4C)平台分子的概念(图3a)。基于2,3-吲哚二甲醇的结构特征,通过调节两个羟基的反应活性,从而控制环加成反应的区域选择性。即:与R1相邻的OH具有较小的空间位阻,因此在B*-H存在下脱水后生成的碳正离子反应性更高,而与两个R相邻的OH具有较大的空间位阻,因此反应性较低。此外,当使用合适的双亲核试剂作为反应物时,亲核性更高的位点会优先进攻与R1相邻的碳正离子。然后,另一个亲核性较低的位点再进攻与两个R相邻的碳正离子,从而控制区域选择性,实现(4+n)环加成反应。在反应过程中,B*-H阴离子会与中间体形成氢键和离子对相互作用,从而控制环加成反应的对映选择性。
基于这一概念,石枫、谭伟等人设计了手性磷酸(CPA)催化下2,3-吲哚二甲醇分别与吲哚和2-萘酚的催化不对称(4+2)和(4+3)环加成反应(图3b),对映选择性构建手性四氢吲哚并咔唑骨架和手性吲哚并七元环骨架。在该设计中,作者考虑到吲哚C3位的亲核性高于C2位,2-萘酚中萘基C1位的亲核性高于酚羟基,这些将有助于控制反应的区域选择性。选择CPA作为高效的手性催化剂,是由于其具有与OH和NH基团形成氢键来活化底物的能力。值得注意的是,高对映选择性构建手性四氢吲哚并咔唑骨架非常具有挑战性,这是由于该类骨架的两个吲哚环只有很小的差异,引入R2基团是为了使该类骨架产生不对称性(手性中心)。由于该手性中心周围具有非常相似的结构,导致难以控制该类骨架的对映体选择性。此外,由于七元环的中环结构中存在不利的熵效应和跨环相互作用,对映选择性构建手性吲哚并七元环骨架也非常具挑战性。
因此,该工作的意义在于不仅将实现2,3-吲哚二甲醇参与的第一例催化不对称(4+n)环加成反应,首次实现吲哚二甲醇作为4C平台分子在不对称催化合成中的应用,而且将首次实现具有挑战性的手性四氢吲哚并咔唑骨架的对映选择性构建。更重要的是,该工作将对反应机理和产物的生物活性进行探究,深化对该类吲哚甲醇特性的理解以及发现手性吲哚并环骨架的潜在应用。
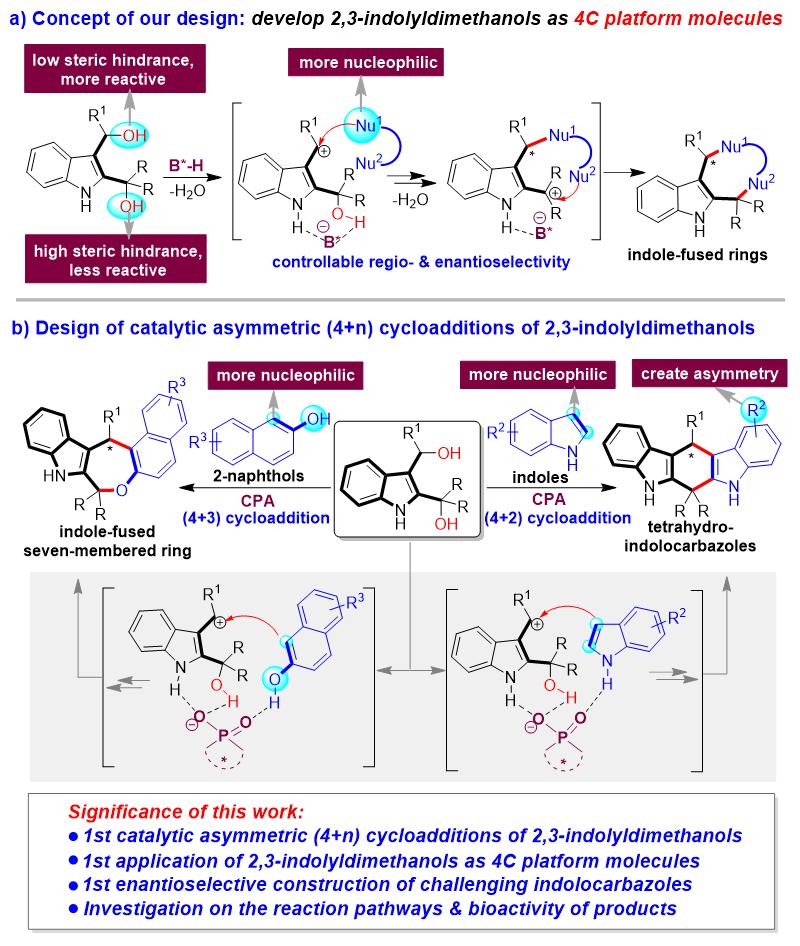
图3. 2,3-吲哚二甲醇参与的区域选择性和对映选择性(4+n)环加成反应的设计
基于上述设计,石枫团队成功地实现了2,3-吲哚二甲醇 1 与吲哚 2 的催化不对称(4+2)环加成反应,高收率(up to 97% yield)、高对映选择性(up to 98% ee)地合成了一系列手性四氢吲哚并咔唑化合物 3 (表1)。值得注意的是,2,3-吲哚二甲醇 1 中的R2/R3基团可以是芳基、杂芳基和烷基,均可以参与催化不对称(4+2)环加成反应,获得高的对映选择性。这在吲哚甲醇化学中非常具有挑战性,因为烷基取代的吲哚甲醇通常显示出非常低的反应性和对映选择性控制能力。所以,烷基取代的2,3-吲哚二甲醇能够参与该反应,高对映选择性地得到产物 3ra-3va ,说明了该(4+2)环加成反应的普适性。更重要的是,一些含有天然产物结构的吲哚 2 也适用于该催化不对称(4+2)环加成反应,以良好的产率和优异的非对映选择性生成天然产物衍生的四氢吲哚并咔唑 3an-3ar ,由此为一些天然产物的后修饰提供了一种有效的方法。值得一提的是,该(4+2)环加成反应具有非常高的区域选择性,只观察到产物 3 的生成,说明该策略可以高效地控制2,3-吲哚二甲醇参与环加成反应的区域选择性。
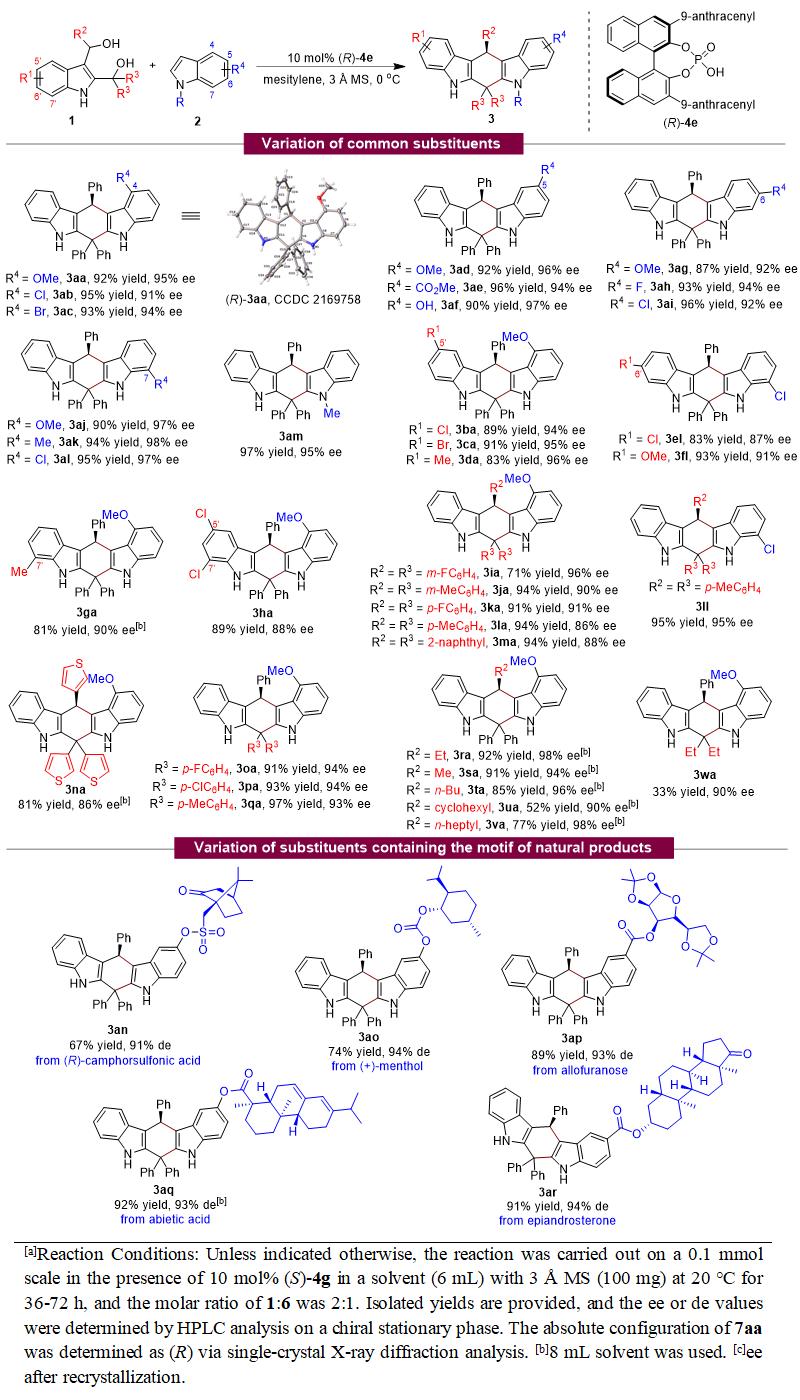
表1. 2,3-吲哚二甲醇参与的催化不对称(4+2)环加成反应的普适性
此外,该团队也实现了2,3-吲哚二甲醇 1 与2-萘酚 6 的催化不对称(4+3)环加成反应,高收率(up to 96% yield)、高对映选择性(up to 97% ee)地合成了一系列手性吲哚并氧杂七元环化合物 7 (表2)。同样,该(4+3)环加成反应也为天然产物的后修饰提供了一种有效的方法,并表现出特定的区域选择性。
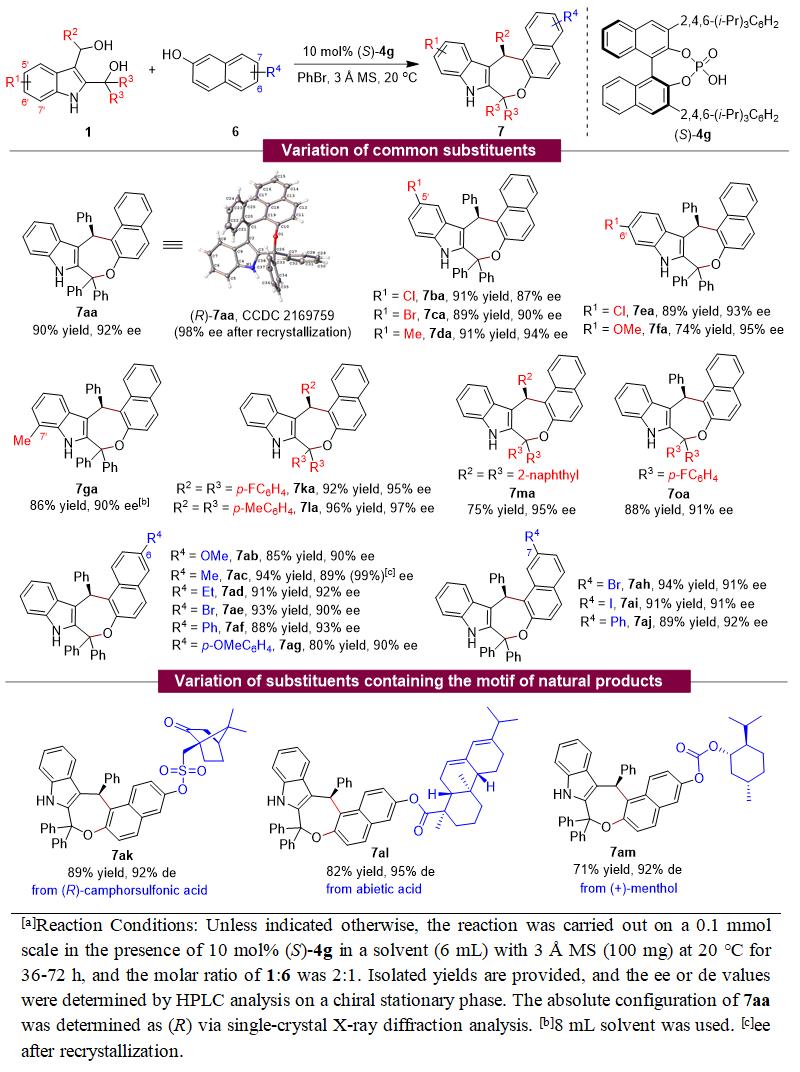
表2. 2,3-吲哚二甲醇参与的催化不对称(4+3)环加成反应的普适性
为了研究2,3-吲哚二甲醇参与的催化不对称(4+n)环加成反应的实用性,作者进行了1 mmol规模的反应和合成转化(图4a-4b)。同时,作者还对合成的一些手性四氢吲哚并咔唑化合物进行了抗肿瘤活性筛选,发现手性产物 3 对PC-3肿瘤细胞显示出很强的细胞毒活性(图4c),其IC50值在6.1-40.6 µM之间。其中,产物 3ac 对PC-3肿瘤细胞具有最强的细胞毒活性,IC50值为6.1 µM。因此,生物活性筛选表明,该类手性四氢吲哚并咔唑在药物化学方面具有潜在的应用前景。
该团队还进行了一些对照实验来探究可能的反应机理和活化模式(图4d-4e)。对照实验表明,2,3-吲哚二甲醇结构中的NH基团在控制催化不对称(4+n)环加成反应的对映选择性或反应活性方面起着至关重要的作用(图4d)。此外,作者发现催化不对称(4+3)环加成过程中存在一定程度的动力学拆分过程,该过程有助于获得高对映选择性的产物 7aa (图4e)。

图4. 反应的实用性、产物的生物活性及对照实验
为了深入理解2,3-吲哚二甲醇参与的催化不对称(4+n)环加成反应的机理和活化模式,石枫团队与汕头大学倪绍飞老师合作,对(4+2)和(4+3)环加成反应的历程和活化模式等进行了理论研究。如图5a所示,在CPA ( R )- 4e 存在下,2,3-吲哚二甲醇 1a 的C3位羟基先脱水,经历过渡态 TS1 得到碳正离子中间体 Int2’ ,其中水分子与CPA ( R )- 4e 阴离子形成氢键。计算结果表明,水分子的释放将导致碳正离子与CPA阴离子之间的氢键相互作用更强,从而产生更稳定的中间体 Int2 。值得注意的是,在 TS1 中,CPA ( R )- 4e 与2,3-吲哚二甲醇的两个羟基都形成了氢键,这意味着C2位的OH基团与催化剂形成氢键作用后,有助于促进C3位OH基团的脱水。脱水后,如 Int3 所示,CPA( R )- 4e 同时与碳正离子中间体和4-甲氧基吲哚 2a 形成多个氢键,通过过渡态 TS2 促进手性中间体 Int4 的形成。然后, Int4 经 TS3 再芳构化生成 Int5 ,再经 TS4 脱水生成碳正离子中间体 Int6 。随后, Int6 经历 TS5 进行分子内加成得到 Int7 ,再通过 TS6 迅速发生吲哚环的芳构化,得到最终产物( R )- 3aa 。此外,为了解释产物( R )- 3aa 绝对构型的产生,作者比较了决定对映选择性的关键过渡态 TS2 和 TS2’ 的能垒(图5b)。结果发现, TS2’ 的吉布斯自由能(-13.7 kcal/mol)高于 TS2 (-16.3 kcal/mol),由此解释了产物( R )- 3aa 绝对构型产生的原因。
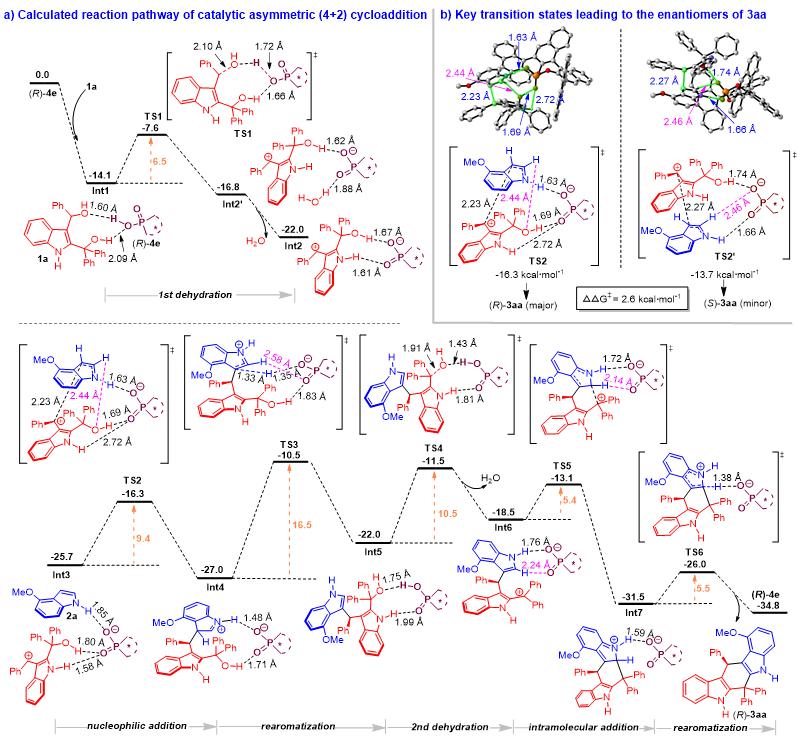
图5. 催化不对称(4+2)环加成反应的DFT计算路径及吉布斯自由能
为了验证碳正离子中间体 Int2 和 Int6 的存在,石枫团队对催化不对称(4+2)环加成进行了动力学研究(即Hammett分析)(图6)。基于测定的具有不同电性取代基(X或Y)的底物参与反应的速率,建立了log(KX/KH)与σp+的Hammett方程。结果发现:log(KX/KH)与σp+的线性关系显著,反应常数 ƿ 均为负值。这表明反应过程中存在正电荷积累的阳离子性质过渡态,从而进一步从实验角度证实了碳正离子中间体 Int2 和 Int6 的形成。
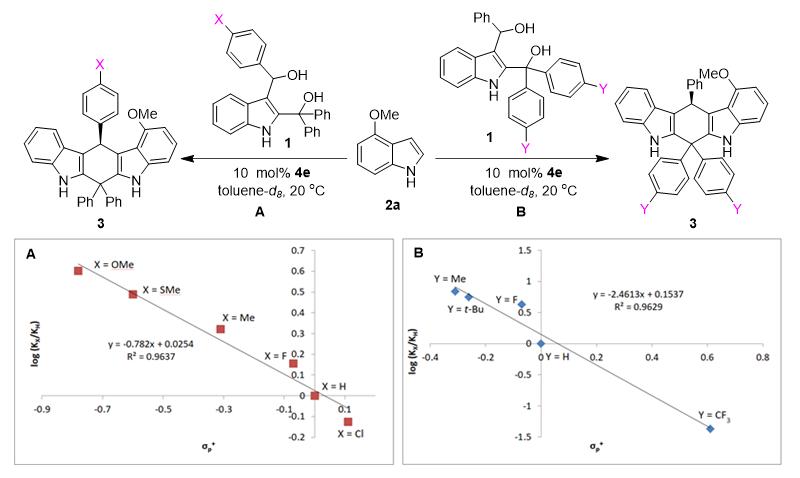
图6. 验证碳正离子存在的动力学研究
此外,作者也对2,3-吲哚二甲醇 1a 与2-萘酚 6a 的催化不对称(4+3)环加成反应的途径进行了DFT计算(图7)。如图7a所示,底物 1a 先脱水,通过 TS7 生成碳正离子中间体 Int9’ ,其中水分子与CPA ( S )- 4g 阴离子形成氢键,水分子的释放产生了更稳定的中间产物 Int9 。与(4+2)环加成类似,在氢键作用下,反应先后经历分子间和分子内亲核加成,最终完成(4+3)环加成反应,得到产物( R )- 7aa 。如图7b所示,作者也比较了决定对映选择性的过渡态 TS8 和 TS8’ 的能垒,发现 TS8 的能垒比 TS8’ 低2.4 kcal/mol,从而解释了产物( R )- 7aa 绝对构型产生的原因。
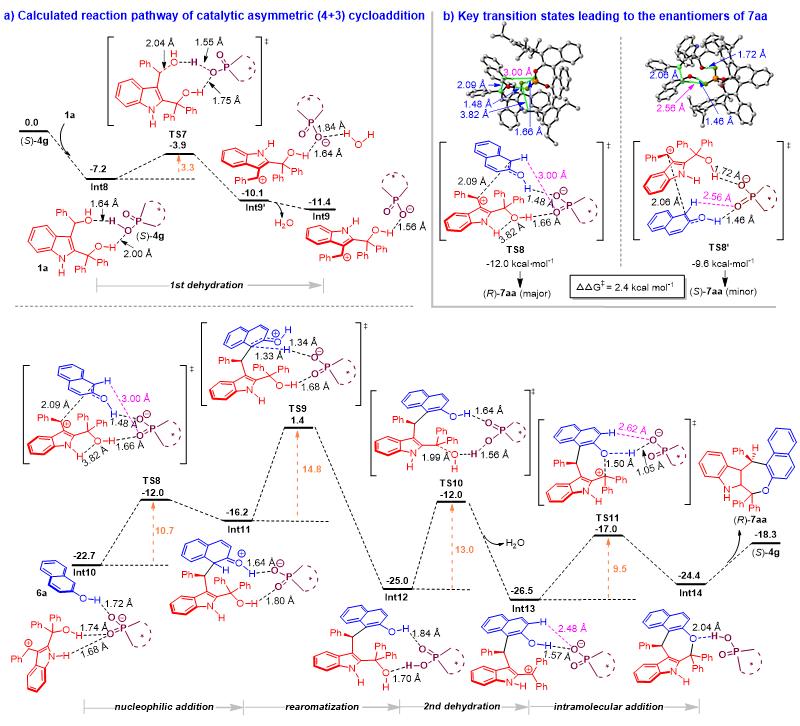
图7. 催化不对称(4+3)环加成反应的DFT计算路径及吉布斯自由能
作者在优化催化不对称(4+2)环加成反应条件的过程中发现:在相同手性磷酸( R )- 4e 的催化下,当*酮丙**作为溶剂时,产物 3aa 的绝对构型可以发生反转,表现出溶剂依赖的产物绝对构型反转现象(图8)。如图8a所示,在( R )- 4e 催化下,在均三甲苯中,底物 1a 与 2a 发生(4+2)环加成反应得到产物 3aa (84% ee);而在*酮丙**中,同样的反应却得到了绝对构型相反的对映体 ent - 3aa (-66% ee)。为了深入探究这一现象,作者采用多种酮类作为溶剂,发现反应在含有 α -氢的酮类溶剂中均生成绝对构型反转的 ent - 3aa (-11% ee到-44% ee)。然而,不含 α -氢的2,2,4-四甲基戊-3-酮作为溶剂时,却不能使 3aa 的绝对构型发生反转。这些结果表明,酮类溶剂的 α -氢在产物绝对构型反转中起着重要作用。在此基础之上,作者通过采用*酮丙**作为溶剂,实现了绝对构型反转的对映体 ent - 3 的合成(图8b)。为了解释 3aa 在*酮丙**中绝对构型反转的原因,作者计算了考虑*酮丙**作用的关键过渡态 TS2a 和 TS2a' 的能垒(图8c),结果发现:由于*酮丙**的羰基和 α -氢都参与了与催化剂和底物的相互作用,使 TS2a’ 的吉布斯自由能(-18.9 kcal/mol)低于 TS2a (-17.1 kcal/mol),从而导致( S )- 3aa 作为主要产物生成。
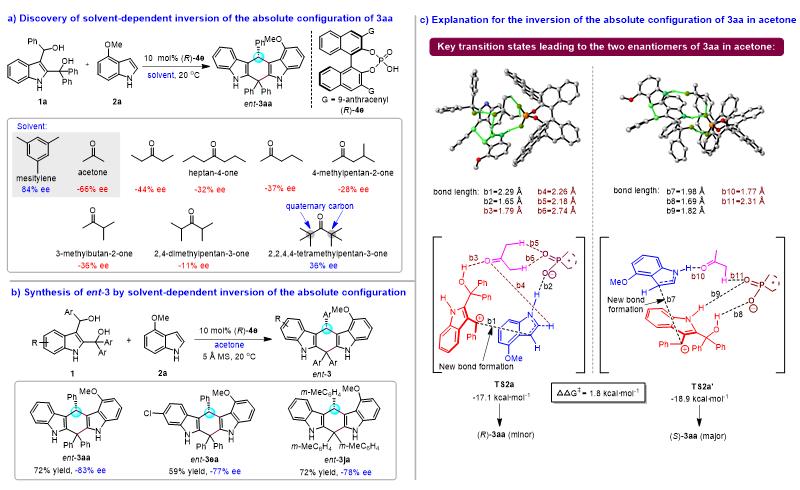
图8. 溶剂依赖的产物绝对构型反转研究
小结
综上所述,江苏师范大学/常州大学石枫、谭伟等人首次实现了2,3-吲哚二甲醇参与的催化不对称(4+2)和(4+3)环加成反应,高收率、高区域选择性和对映选择性地构建了多种手性吲哚并环骨架。该工作不仅代表了第一例吲哚二甲醇参与的催化不对称环加成反应,而且首次将吲哚二甲醇作为4C平台分子应用于不对称催化合成。此外,该方法首次实现了具有挑战性的手性四氢吲哚并咔唑骨架的对映选择性构建,并发现了一些具有显著抗肿瘤活性的手性四氢吲哚并咔唑分子。此外,通过实验与理论计算相结合,探究了2,3-吲哚二甲醇参与的催化不对称(4+2)和(4+3)环加成反应的途径、中间体和活化模式,为进一步理解该类新型吲哚甲醇的反应特性提供了理论基础。因此,该研究不仅解决了吲哚二甲醇参与的催化不对称环加成反应这一未知化学中的挑战性问题,而且为高对映选择性构建手性吲哚并环骨架提供了高效的策略,将有力促进吲哚甲醇化学的发展。
上述研究成果发表在 Angew. Chem. Int. Ed. (DOI: 10.1002/anie.202305450),江苏师范大学硕士研究生 张家毅 (实验部分)和汕头大学本科生 陈嘉仪 (理论计算部分)为共同第一作者,江苏师范大学/常州大学 石枫 教授、江苏师范大学 谭伟 副教授、汕头大学 倪绍飞 老师为共同通讯作者。该工作的实验部分由 谭伟 副教授和 石枫 教授共同指导完成,理论计算部分由 倪绍飞 老师指导并与石枫教授探讨完成。该项工作主要得到了国家自然科学基金和江苏省自然科学基金的资助。
Asymmetric (4+n) Cycloadditions of Indolyldimethanols for the Synthesis of Enantioenriched Indole-Fused Rings
Jia-Yi Zhang, Jia-Yi Chen, Cong-Hui Gao, Lei Yu, Shao-Fei Ni, Wei Tan, Feng Shi
Angew. Chem. Int. Ed ., 2023 , DOI: 10.1002/anie.202305450
通讯作者简介
石枫 教授

石枫,江苏师范大学教授、常州大学特聘教授。主要从事催化不对称合成手性杂环的研究,聚焦手性吲哚化学这一研究领域,为构建结构复杂多样的手性杂环骨架提供了高效、高选择性的方法。以通讯作者在《 Acc. Chem. Res. 》、《 Angew. Chem. Int. Ed. 》、《 J. Am. Chem. Soc. 》、《 Fundam. Res .》、《 Sci. China Chem. 》、《 Chin. J. Chem. 》等业界公认的国际重要科技期刊发表学术论文130余篇,获得发明专利授权8项,入选Elsevier 2020-2022高被引学者。荣获江苏省第二届“十佳研究生导师”提名奖、江苏省研究生教育改革成果奖优秀奖、江苏省科学技术二等奖、教育部自然科学二等奖、Thieme Chemistry Journal Award、Asian Core Program Lectureship Award、新和成《中国化学》创新奖等奖项。担任《有机化学》、《 Chem. Synth .》编委、《 Eur. J. Org. Chem. 》编委会主席团成员、《 Org. Chem. Front. 》、《 J. Org. Chem. 》和《 Synthesis 》国际编委。
谭伟 副教授

谭伟,江苏师范大学化学与材料科学学院副教授。2015年于江苏师范大学获得硕士学位(导师:石枫教授),2019年于华东师范大学获得博士学位(导师:姜雪峰教授),同年被引进到江苏师范大学。主要从事生物活性手性杂环分子的催化不对称合成、轴手性杂环骨架的催化不对称构建研究,近年来以通讯作者或第一作者在《 Fundam. Res. 》、《 J. Am. Chem. Soc. 》、《 Angew. Chem. Int. Ed. 》、《 Sci. China Chem. 》、《 Chin. J. Chem .》、《 Org. Lett. 》、《 Chem. Commun .》等业界公认的国际重要科技期刊发表学术论文20余篇。
倪绍飞 博士

倪绍飞博士,现入职汕头大学化学系和广东省有序结构材料的制备与应用重点实验室。2019年于西班牙加泰罗尼亚化学研究所(ICIQ)获得博士学位(导师:Prof. Feliu Maseras),期间获欧盟Erasmus+奖学金项目资助,赴瑞典斯德哥尔摩大学有机化学系访问学习(导师:Prof. Fahmi Himo)。2020年在德国哥廷根大学从事博士后研究工作(导师:Prof. Lutz Ackermann)。2020年9月入职汕头大学。主要从事计算化学,包括金属有机化学、有机反应机理、催化反应机理及发光材料的光激发电子跃迁机理等领域的研究,共发表SCI论文50多篇,近三年以第一作者或者共同通讯作者在《 Coord. Chem. Rev .》、《 Angew. Chem. Int. Ed .》、《 Adv. Funct. Mater .》、《 ACS Catal. 》、《 Green Chem .》、《 Chem. Sci. 》、《 Chin. J. Chem .》等国际知名期刊发表学术论文10余篇。
石枫团队网站:
https://www.x-mol.com/groups/Shi_Feng