本文来自微信公众号:X-MOLNews
杂环化合物广泛存在于药物分子、天然产物以及农用化学品等领域中,其中哌啶是含氮杂环药物中的关键结构片段。在过去的几十年里,化学家通过位点选择性的策略对哌啶外围进行*能官**团化以获得新的化学结构和功能(图1A),但是对核心骨架重新构建的研究却少之又少。与碳环的收缩方法相比,哌啶缩环的方法相对较少。目前,有关哌啶甚至是饱和氮杂环的缩环策略也有些许报道,例如:1)双环季铵阳离子中间体与外部亲核试剂进行的缩环反应(图1B);2)氧化C( sp3 )-N键裂解的缩环反应(图1B);3)α-羧基四氢异喹啉(THIQ)及其衍生物的异常重排(即内环到外环的氮原子移位,图1C);4)光介导的缩环反应(图1D)等等。尽管如此,但上述方法通常存在反应条件苛刻、底物局限、*能官**团兼容性不好等缺点,因此极大地限制了底物结构的多样性和反应的选择性,严重地阻碍了药物结构的后期修饰。
受到上述策略的启发,美国 加州大学伯克利分校 的 Richmond Sarpong 教授课题组 通过Norrish II型反应,成功地实现了可见光介导的α-酰化饱和杂环的缩环反应 (图1E),构建了一系列 cis -1,2-二取代环戊烷骨架,并将该方法扩展到其它饱和杂环(如吗啉、四氢吡喃、四氢异喹啉和噻烷)、药物衍生物、肽和糖的后期重构。相关成果发表在 Science 上,美国默克(Merck & Co. Inc.)的 Yu-hong Lam 与 Charles S. Yeung 为论文的共同通讯作者。

图1. 哌啶多样化的途径。图片来源: Science
如图2A所示,作者设想在辐照后,α-酰化前体(如 I )中芳酰基处于pseudo-axial,从而避免烯丙基张力(A1,3-like strain)的作用,经历激发和系间窜越(ISC)过程获得三线态中间体 II ,后者经Norrish Type II型 1,5-氢原子转移(HAT)产生相应的1,4-双自由基中间体 III ,随后 III 发生 C-N 键裂解产生亚胺-烯醇 IV ,最后经分子内Mannich反应便可构建环戊烷骨架。然而,在光催化下会有其它副反应发生(图2B),例如:1)反应产物 V 形成激发态 VI ;2)Norrish I型过程;3)Norrish-Yang环化过程等等。
尽管存在副反应和过度反应的挑战,但基于 I 和 V 吸光率的差异,作者希望通过调节光的波长选择性地控制反应性,从而最大限度地减少副反应。经过初步的研究,作者发现光介导的缩环反应在400 nm波长下效果最优,能以73%的收率得到1,2-二取代环戊烷 4a (图2C);而较长(450 nm)或者较短波长(385 nm)的光源均不利于该反应,甚至不反应。进一步的研究表明:1)哌啶氮上磺酰基具有独特的反应性,是其它吸电子基团(如Bz、Boc或Piv)不能比拟的;2)溶剂同样对反应很重要,苯是最佳的反应溶剂。这是因为磺酰基与苯可以增强 4a 的分子内氢键,从而提高产物 4a 的稳定性;3)降低底物浓度可以减慢副反应的竞争速度以达到提高产率的目的;4)最优条件下与流动化学相结合可以在克级规模上进行反应。不过,含有其它保护基团的底物(如酰胺和氨基甲酸酯底物)反应性却较低,为此作者选择酰胺保护底物 3n 为模板底物,筛选了 46 种已知的光敏剂和光催化剂,最终发现3-Cyanoumbelliferone( IX )作为有效的添加剂可提高反应效率。
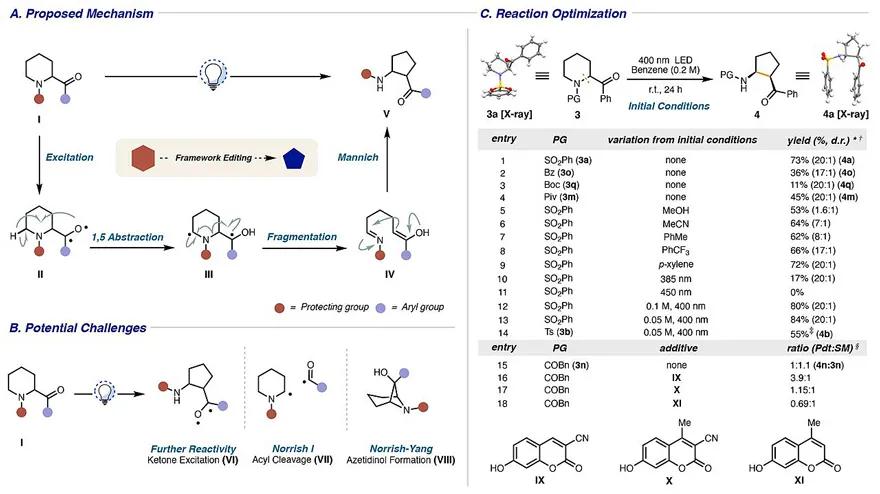
图2. 反应条件优化。图片来源: Science
在最优条件下,作者对底物范围进行了考察(图3)。首先,他们考察了氮原子上的不同基团(图3A),结果显示无论芳基磺酰基哌啶类化合物带有吸电子基团如酮( 3h )、酯基( 3i )、卤素原子( 3e-3g ),还是给电子基团如醚( 3c 、 3d ),都能以中等至良好的收率获得相应的缩环产物。值得一提的是,在cyanoumbelliferone( IX )的存在下,哌啶氮上也可以耐受酰基( 3m-3p )、氨甲酰基( 3q )、脲基 ( 3r、3s )和亚磷酰胺( 3t、3u )。其次,作者对α-芳酰基的底物范围进行了研究(图3B),结果显示空间位阻对该反应的影响很大,例如:对甲基取代的底物( 5a )比邻甲基取代的底物( 5b )反应效果(收率和非对映选择性)更好。另外,该反应还能耐受多种*能官**团(如醚( 5c )、硫醚( 5d )、氟原子( 5e )、氰基( 5f )、三氟甲基( 5g ))。需要指出的是,萘基酮( 5h、5i )、噻吩酮( 5k ))甚至非芳基酮( 5l-5n )都能实现这一转化,以中等至良好的收率得到所需产物。最后,作者探究了取代哌啶和其它饱和杂环的底物范围(图3C),结果显示γ-单取代哌啶 ( 7a )、四氢异喹啉( 7b )、吗啉( 7c )、α-酰化噻烷( 7d )以及四氢吡喃(THP)衍生物( 7e )均具有很好的兼容性,以中等至较好的收率和非对映选择性获得相应的环戊烷衍生物。
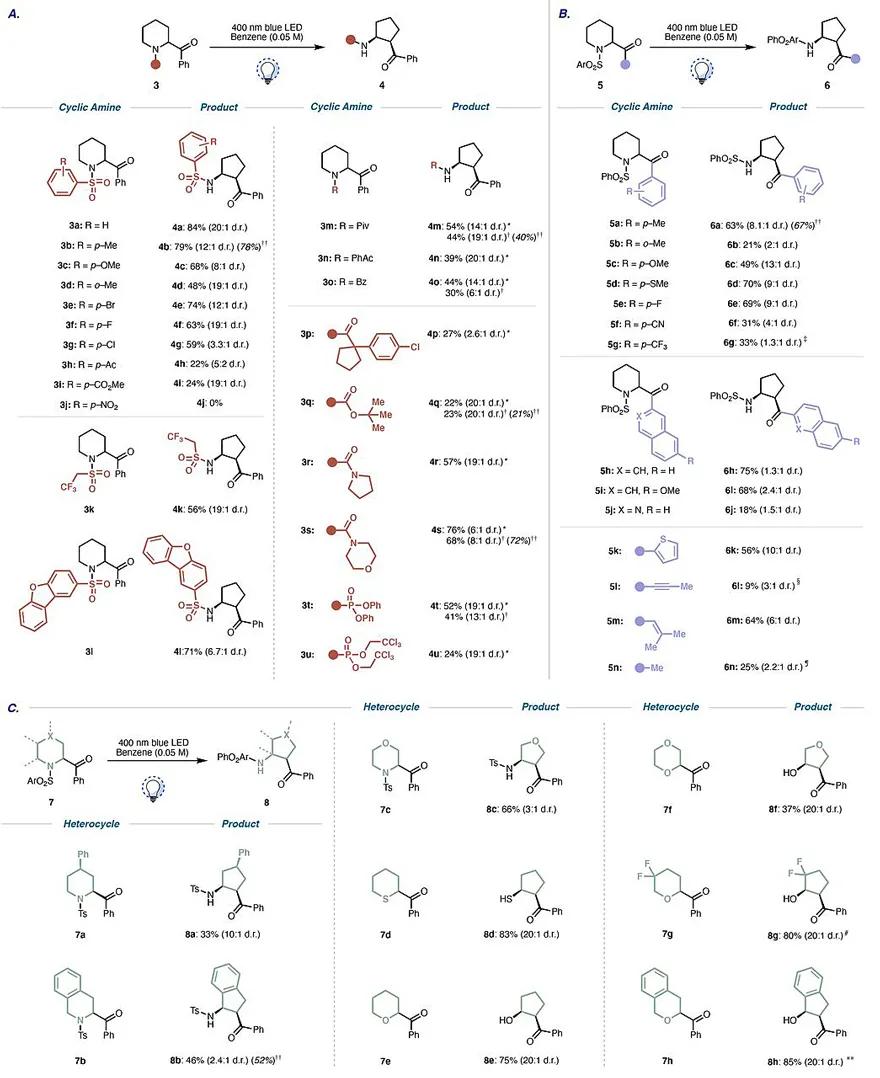
图3. 底物拓展。图片来源: Science
接下来,作者将环收缩方法应用于生物活性小分子的构建中,以证明该方法在药物后期衍生化的潜力。如图4A所示,MDMC( 9a ,兴奋剂)、Rimitrol( 9b ,支气管扩张剂)、Cathone( 9c ,DAT*制剂抑**)和Mefloquine( 9d ,抗疟药)衍生物均可顺利地发生缩环反应,以中等的收率获得相应的环戊烷异构体。此外,作者还在肽多样化(图4B)与糖化学的编辑中(图4C)中利用了缩环转换,如含有甘氨酸的肽( 11 )被转化为相应的氨基环戊烷( 12 )、 D -半乳糖衍生的双*酮丙**化物( 13 ) 产生开环的产物( 14 ),进一步体现了该方法在多肽修饰以及多糖编辑中具有强大的生命力。
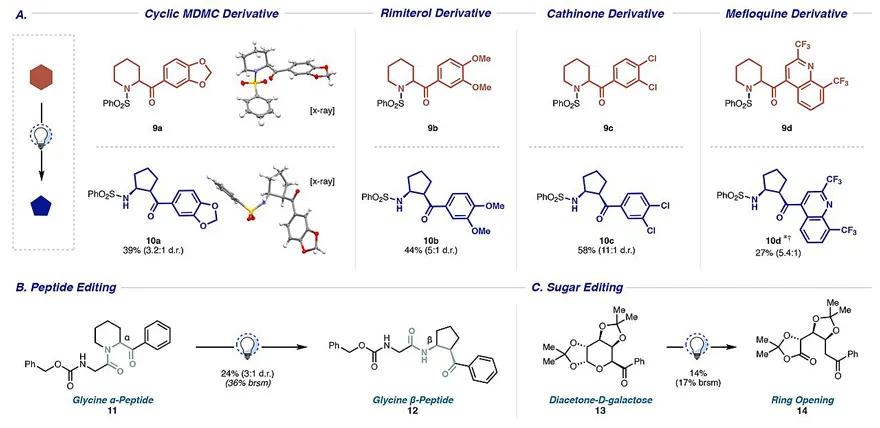
图4. 活性物质的制备。图片来源: Science
为了进一步探究反应机理,作者对 N -甲苯磺酰基哌啶衍生物 3b 的反应进行了计算研究(图6A)。最初,作者认为1,5-HAT过程的位置选择性可归因于α-氨基氢原子的强酸性和低键解离能( 33b → 3A )。事实上,DFT计算表明哌啶 α-位氢原子的HAT过渡态能量比 γ 位C-H键活化的能量低9.0 kcal/mol;同时还揭示了过渡态中N-S键的最佳构象(即羰基和磺酰氧基之间保持最大分离)。其次,作者推断 cis -氨基环戊烷(如 4b )的非对映选择性可能是由Mannich型环闭合过渡态中的一系列非共价相互作用(如π-堆积和氢键)引起的。DFT计算的过渡态结构支持 Mannich 型环化/C-C 键的形成与烯醇的部分质子转移到 N-tosyl基团的过程相一致。最后,作者通过三种设想来支持实验中所观察到的非对映选择性,其中最有可能的情况是双自由基的寿命足够长且酰基键可旋转。在此情况下,只有 ( E/Z )和( E/E )亚胺-烯醇是可用的,并且 TS-1-4 之间的能量差异将决定立体化学结果(计算与实验结果相一致)。然而, cis -非对映选择性主要源于 TS-3 和 TS-4 之间的能量差异,并且可以通过更短且更强的氢键以及在 TS-3 中形成 C-C 键的取代基的交错排列使其趋于合理化(图5A、 1B→14b )。值得注意的是, N -甲苯磺酰基哌啶 3b 向任一环戊烷产物的整体转化都是放热的。
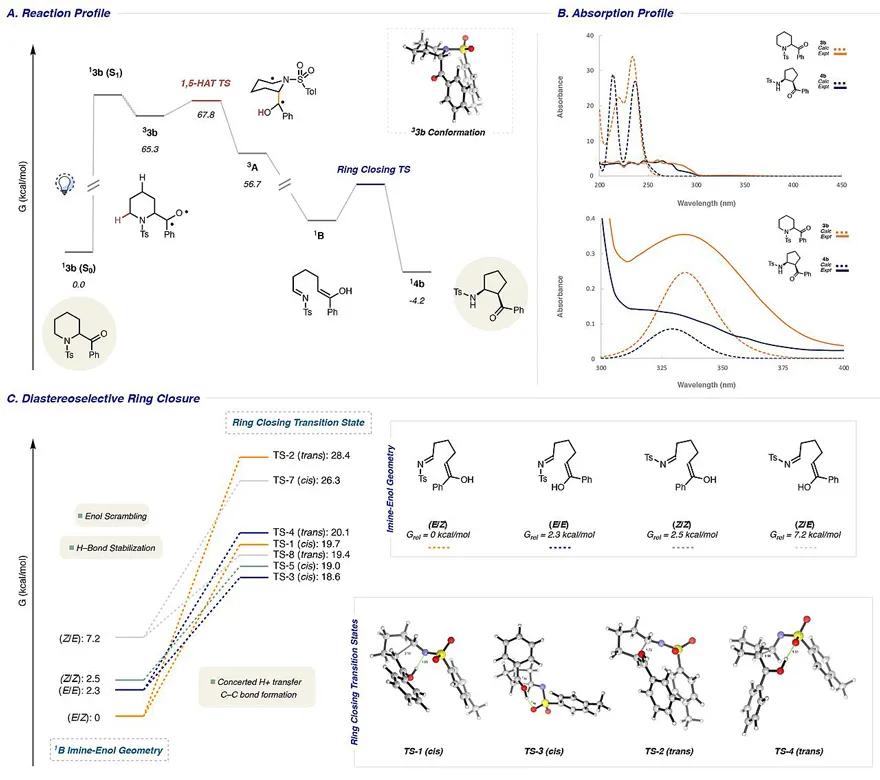
图5. 计算化学研究。图片来源: Science
最后,作者设想能否在亚胺质子化与烯醇去质子化后提高Mannich步骤速率,从而有利于过渡态被进攻以获得单一的对映体。在此情况下,氢键和离子配对的结合有望产生手性纯的Mannich产物(图6)。为此,作者以手性磷酸( R )-TRIP ( CPA1 )为添加剂,以76%的收率、20:1的d.r.值以及84%的e.e.值获得相应的目标产物(-)- 4b ;使用SPINOL-衍生的磷酸( R )-XYL-SPA( CPA2 )为添加剂,以89%的收率、20:1的d.r.值以及90%的e.e.值获得相应的目标产物(+)- 4b 。需要指出的是,酰胺、尿素、THIQ、吗啉以及THP衍生物都能兼容该反应,以中等至较好的收率和选择性制备了一系列手性环戊烷( 4m、4s、8b、8c、8e、8g )。
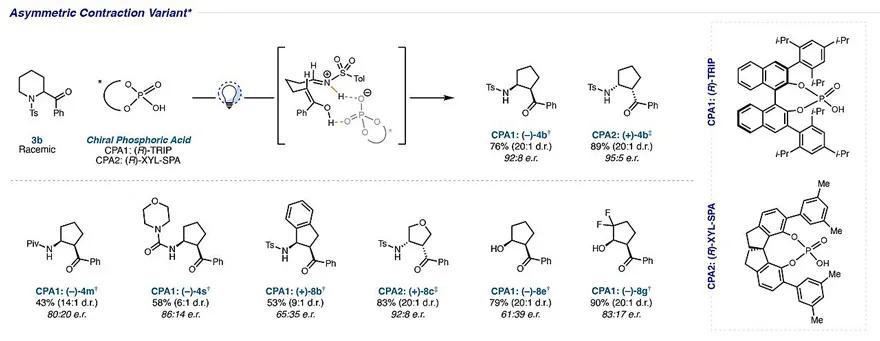
图6. 不对称缩环反应。图片来源: Science
总结
Richmond Sarpong教授课题组通过Norrish II型反应,成功地实现了可见光介导的α-酰化饱和杂环的缩环反应,构建了一系列 cis -1,2-二取代环戊烷骨架,并将该方法扩展到其它饱和杂环、药物衍生物、肽和糖的后期重构以及手性环戊烷的构建中。基于计算化学和实验结果,反应的机理和非对映选择性的起源得到了进一步的解释。
Photo-mediated ring contraction of saturated heterocycles
Justin Jurczyk, Michaelyn C. Lux, Donovon Adpressa, Sojung F. Kim, Yu-hong Lam, Charles S. Yeung, Richmond Sarpong
Science , 2021 , DOI: 10.1126/science.abi7183
导师介绍
Richmond Sarpong
https://www.x-mol.com/university/faculty/32
(本文由 吡哆醛 供稿)