我们生活在一个以多样性为特征的社会中,无论是身体形态、思维方式,甚至是生理构造,都有着各种各样的差异;在我们身边,有一类特殊的群体,民间将她们归类为"石女"。
石女虽然较罕见,但仍影响着全球数以万计的女性。据了解, 全球石女的发病率大约是1/5000到1/7000,其中包括因为先天畸形、外伤或者手术等原因导致的后天石女。 虽然发病率并不算高,但对于每一个受影响的女性来说,这无疑是个人生的重大打击。
许多患者可能由于羞耻感或对情况不理解而不愿意寻求帮助,这就导致她们无法倾诉苦痛,让人倍感心酸。

一、所谓石女,究竟和正常女性之间有何异同?
在进行深入讨论前,首先要明确“石女”的定义。
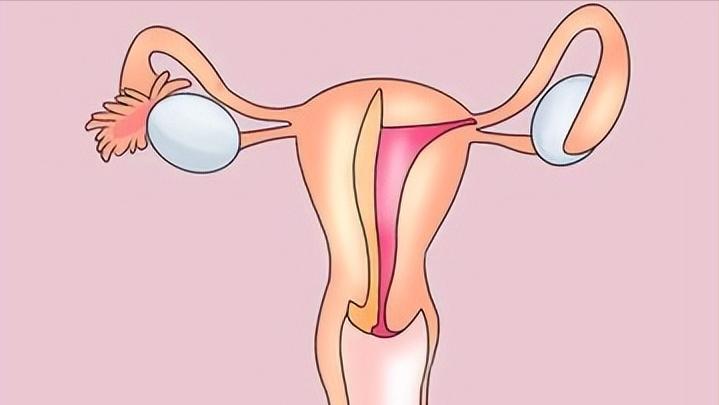
“石女”通常是指那些由于先天性身体结构问题,无法进行正常婚姻生活乃至无法生育的女性。 从医学角度看来,“石女”是指那些由于某种原因,在胚胎发育阶段没有形成完整的内生殖器官(尤其是阴道和子宫)的女性。当然,正如上文提到的,一些由于继发原因丢失内生殖器官的女性也囊括在列!
通常情况下,这部分女性的体型、面部特征、第二性征如乳房、阴毛等都与正常女性无异,甚至比较美丽。但由于内部生殖系统缺陷,使得她们无法经历月经周期,也就没有生育能力。

下面我们分别从几个方面来详细说明:
1. 保持正常女性的第二性征
石女,在患者中表现为阴道、子宫或两者共同的缺陷甚至缺失。但 患者的外部生殖器和其他体征,如第二性征(乳房发育、正常体毛分布等)与非患者无异,这主要源于雌激素的作用。
雌激素主要来源于卵巢,其在发育过程中起着关键的作用。
雌激素可以促进女性乳房的发育,加强皮肤和其他组织的血管供应,改变体毛分布,并且对骨骼的生长和成熟也有影响。
因此,即便石女患者的内部生殖器官未能正常发育,由于卵巢功能正常并产生了雌激素,她们的第二性征(乳房发育、正常体毛分布等)与正常女性无异。

2. 无法经历正常的月经周期
患者的子宫和阴道可能部分或全部缺失,这导致她们无法经历正常的月经周期。
在一个典型的月经周期中,子宫内膜会在雌激素和孕酮的影响下逐渐增厚,准备接受可能会来临的受精卵。然而,如果这个过程中没有受精卵着床,那么子宫内膜就会脱落并通过阴道流出。 对于石女来说,由于缺乏必要的器官结构,这一周期性过程无法进行。
3. 生育能力的丧失
由于石女患者的子宫或阴道可能部分或完全缺失,她们无法完成正常的婚姻生活,也无法进行自然的受精和怀孕过程,因此无法生育。
虽然他们的卵巢通常能正常工作并产生卵子,但由于往往没有合适的地方供胚胎发育,所以不能实现正常的孕育过程。
4.性别认知的混乱
石女症状对患者的影响并不仅限于生物学层面,还包括心理社会层面。
这种病症通常在青春期的时候被发现,这是一个自我认知和形象正在发展的关键阶段,因此可能对她们的身份感、自尊和性别角色感产生重大影响。
在发现自己无法经历月经、进行婚姻生活或自然怀孕后,许多患者会有严重的焦虑、抑郁和自我价值缺失感。这可能进一步影响她们的人际关系、社交活动和未来规划。
因此,正是由于这些不同,导致了石女虽然外表可能和正常女性无异,但其实身心都受到很大影响。然而,你知道吗?石女并不完全一样,甚至有真假之分!

二、石女之间也不甚相同?竟还有真假之分?
尽管被统称为“石女”,但实际上,每个石女的具体情况可能会有很大的差异。医学界将其进一步细分为“真石女”和“假石女”。
关于“石女”,广义来说,包括了先天性无阴道或子宫发育不良等情况。
然而,在临床实践中,医生发现并非所有表面看起来像“石女”的患者都是真正的石女。 有些女性虽然生殖器官外貌看起来像“石女”,但其实内部器官比较完整,只是因为一些原因使阴道闭锁或者狭窄,经过手术或者治疗后能恢复正常功能,这被称为“假石女”。
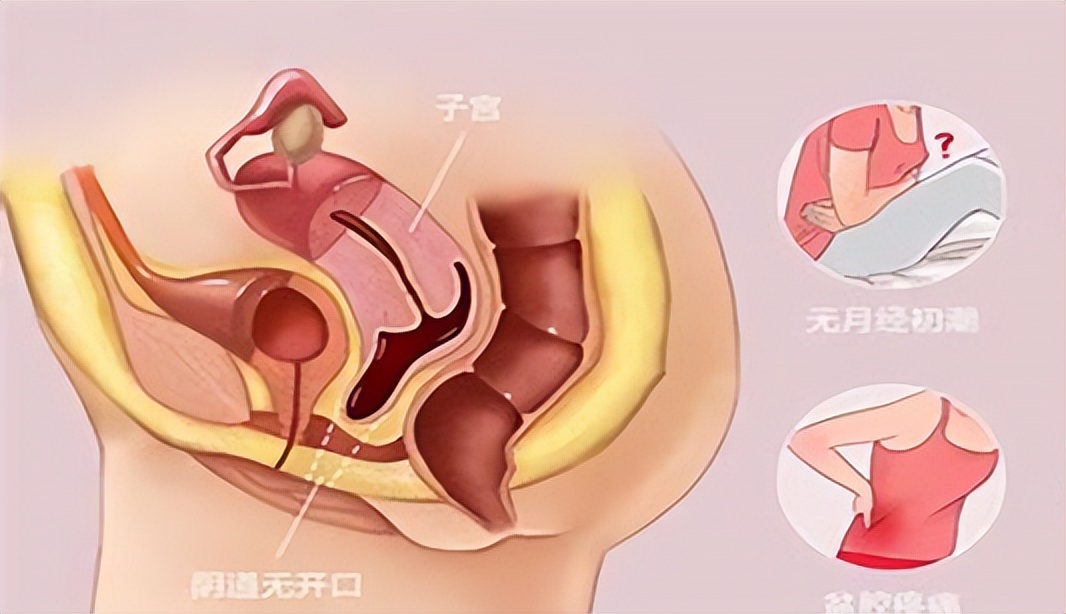
在门诊我们偶尔可以看到年轻女性、新婚夫妻因无法进行夫妻生活而前来就医,这大部分都是先天性阴道闭锁所致。
先天性阴道闭锁通常是由于胚胎发育过程中阴道管发育异常引起的。 最常见的原因是阴道管的上端未能正常开口,形成了一层膜或隔膜,导致阴道的完全或部分闭塞。
事实上,她们是相对幸运的,在经过手术后她们即与正常女性相差不大,并且大部分治疗相对容易一些。当然也有少数患者是阴道缺失,她们只能与真石女一样进行阴道重建,才能恢复功能。
在讲完“假石女”后,我们再来看看何为“真石女”。
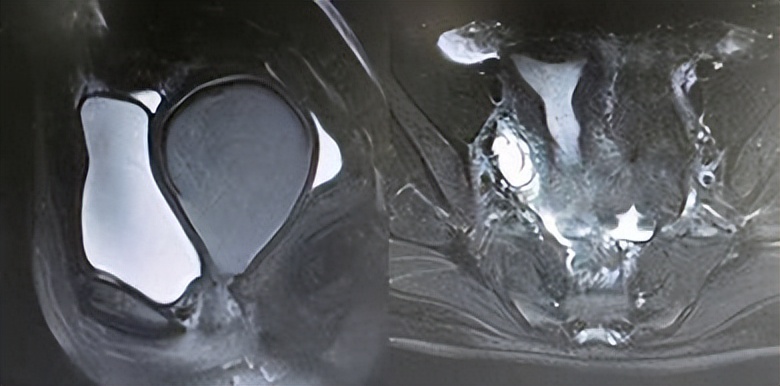
“真石女”通常指的是先天性无阴道或者阴道闭锁且无成熟子宫的女性。她们的子宫是完全不存在或者发育不良,因此无法从医学意义上得到真正的恢复。
在医学角度上,“真石女”的病情主要表现为先天性阴道闭锁、阴道过窄或完全无阴道,子宫、卵巢缺失。这一般与以下两种疾病有关:①Mayer-Rokitansky-Küster-Hauser (MRKH) 综合症;②Androgen Insensitivity Syndrome (AIS)。
1.MRKH综合征
大部分“真石女”往往患有一种叫做MRKH综合征的疾病,即先天性无子宫、无阴道,这是一种先天性结构异常,在目前的医疗水平下,暂无法使其恢复。
MRKH综合征的病因尚未完全明确,但目前认为它主要与胚胎发育过程中苗勒管的异常发育有关。
在胚胎发育过程中,苗勒管是形成女性生殖系统的重要结构,它会分化为子宫、输卵管和阴道。对于患有MRKH综合征的女性来说,可能是苗勒管的发育出现了问题,导致子宫或阴道的缺失或发育不良。
还有一些研究表明,MRKH综合征可能与某些基因的突变有关。
最近研究者发现了一种名为WNT4的基因突变。这种基因对于女性生殖器官的发育极为关键,如果出现突变,可能会导致石女症状。这些基因在子宫和阴道发育过程中起着重要作用。然而,具体影响MRKH综合征的基因突变仍未完全阐明,并且可能存在多个基因的相互作用。
环境因素也被认为在MRKH综合征的发生中可能发挥一定作用。 例如,母体在怀孕期间暴露于某些药物、化学物质或辐射等有害因素可能会增加患病风险,但具体的环境因素尚不清楚。
2.AIS综合征
少数病人表现出“真石女”的症状,但却并不是MRKH综合征患者,而是雄激素不敏感综合征(AIS)的患者。 在经过染色体检查前往往以为自己是女性,但实际上她们在生物学意义上是男性,只是由于对雄激素不敏感而表现出女性的第二性征,丧失男性的第二性征,甚至由于生殖器官发育不良,不能表现出明显的第一性征。
显而易见,她们无法生育,并且由于雄激素的作用,她们也没有完整的女性性征,故而表现为“石女”。
AIS是一种常染色体显性遗传疾病, 主要影响男性的性发育和性别确定。
AIS 综合征通常由 X 性染色体上的 AR 基因(雄激素受体基因)的突变导致。AR 基因突变会导致雄激素受体蛋白的异常或缺失,影响雄激素受体的结构或功能,使其无法正常地与雄激素相互作用。
这可能包括雄激素受体蛋白结构上的变化、受体信号转导途径的改变,或者受体在细胞核中的定位问题。这些分子缺陷会干扰细胞对雄激素的正常反应,这意味着即使机体产生足够的雄激素,细胞却无法“意识”到指令,使得发育过程失控,从而导致一系列的性发育异常。
需要强调的是,“真”与“假”的区分并没有好坏之分,它只是描述了患者的身体条件。每一种情况都需要专业的医疗关注和治疗,需要社会的同情与关照。

三、石女怎么办?还能正常生活吗?
对于石女来说,想要让其恢复,一般只需将狭窄或闭锁的阴道恢复即可。在临床治疗上,我们一般使用这两种方法来重建或恢复阴道:
1.阴道成形术(Vaginal creation surgery): 这是通过手术创造一个功能性阴道的方法,通常使用组织移植或人工材料来建立阴道结构。手术后,患者需要进行术后恢复和阴道扩张的训练。
2.动态阴道伸展器(Dynamic vaginal dilators): 这是一种非手术的方法,通过使用逐渐增大尺寸的阴道扩张器来保持和延长阴道的长度。患者需要定期使用阴道伸展器来维持阴道的可用性。
当阴道重建完成后,“假石女”由于具有子宫,仍然可以通过自然受孕生育子女。虽然“真石女”因缺少子宫而无法生育,但是生育并不是女性唯一的事情,石女仍然可以以自己的方式的度过人生。

四、写在最后
虽然石女的生理问题可能带来一些困难,比如疼痛、无法进行婚姻生活和自然生育等,但并不代表她们不能过上正常的生活。
随着医疗技术的进步,石女现在可以通过手术来修复阴道,改善婚姻生活和生育的可能性。最重要的是,我们需要理解和关心石女,消除社会对她们的误解和歧视。
只有在理解和接纳的环境中,石女才能够自信地面对生活,寻找自己的幸福。总的来说,“石女”并不是一种标签,而是一种生理状态。尽管她们面临着一些特殊的挑战,但她们依然可以像其他女性一样,追求并享受生活。
参考文献:
[1] Rall K, et al. Mayer-Rokitansky-Küster-Hauser syndrome discordance in monozygotic twins: Matrix metalloproteinase 14, low-density lipoprotein receptor-related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific mosaicism. Fertil Steril. 2015;103(2):494-502.e3.
[2] Hughes IA, et al. Androgen insensitivity syndrome. Lancet. 2012 Dec 22;380(9851):1419-28.
[3] Biason-Lauber A, Konrad D, Meyer M, DeBeaufort C, Schoenle EJ. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. American Journal of Human Genetics. 2009 May;84(5):658-63.
[4] Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertility and sterility. 2011 Mar 15;95(5):1589-94
#秋日生活打卡季#