在阅读文章前,辛苦您点下“关注”,方便讨论和分享。作者定会不负众望,按时按量创作出更优质的内容。
最新的病毒分类第八次报告仅将过去定义的Ⅰ群禽腺病毒(fowl adenovirus,FAdV)归为FAdV属,包括了传统的FAd V及分离自鸡、火鸡、鹅和其它禽类的腺病毒。
由于FAdV多数呈隐性感染,不引起明显临床症状,在免疫学上不仅可诱导感染鸡产生高水平的中和性循环抗体,还可较好的诱导机体的黏膜免疫,是构建载体活疫苗良好的候选病毒。
近几年来,利用各型FAdV构建载体已成为禽病疫苗研究的热点,在国外已有广研究,但离临床应用还有较大距离,国内在此方面的研究较少。本研究利用真核细胞内同源重组的方法构建了表达绿色荧光蛋白的重组禽腺病毒,期望为今后开展禽类活载体疫苗研究提供一个有效的病毒载体。

1型禽腺病毒AV208株(FAdV-1 AV208):中国兽医药品监察所产品;鸡肝癌细胞系(LMH):美国菌种保藏中心产品;DMEM/F12培养液、胎牛血清、抗生素、胰蛋白酶等:Gibco公司产品;pUC18载体、内切酶等:大连宝生物工程公司产品;质粒p EGFP-N1:Clontech公司产品;质粒提取试剂盒:Omega公司产品;Lipofetamine2000转染试剂:Invitrogen公司产品。
在探索FAdV-1 AV208株的复制非必需区时,参考了FAdV-1代表株CELOV的基因组序列(序列号U46 933),其右末端存在着较大的复制非必需区.设计两对引物扩增FAdV-1AV208株包含欲缺失的复制非必需区及侧翼序列的基因组大片段(对应CELOV基因组位置为nt39 294~nt43 747)。

引物对39294U24和43404L23用于扩增nt39 294~nt43 404之间的4 111 bp片段,引物对43309U21和43747L21用于扩增nt43 309~nt43 747之间的438 bp片段,产物送上海英骏生物技术有限公司测序,并与CELOV基因组进行同源比对分析.引物对序列如下:

根据上述基因组大片段的测序结果,分别选取nt39 294~nt40 064及nt43 045~nt43 747的序列片段作为欲缺失复制非必需区的左右侧翼序列片段L和R,也即转移载体的左右同源重组臂.设计两对引物,LU/LL用于扩增左臂,RU/RL用于扩增右臂,产物送INVITROGEN公司测序,并与基因组大片段的测序结果进行比对分析。引物序列如下:

P C R扩增得到的左、右同源重组臂分别经EcoRI/SpeI和SpeI/HindⅢ双酶切后,与经EcoRI/HindⅢ双酶切处理的pUC18质粒相连接,这样同源重组臂之间通过共有的SpeⅠ位点相连接,缺失了两者之间的2 980 bp欲缺失的复制非必需序列,构建了携带左右同源重组臂的中间载体pUC-L-R。连接产物转化大肠杆菌感受态,提质粒后送上海英骏生物技术有限公司测序。
根据pe GFP-N1载体序列信息,设计引物对EU/EL扩增包括hCMV、MCS、eGFP、SV40早期转录polyA信号在内的1 602 bp的片段,产物送上海英骏生物技术有限公司测序,并与peGFP-N1载体序列进行比对分析。得到的序列正确的T载体命名为pMD-eGFP.引物序列如下:

将pMD-eGFP载体与pUC-L-R载体同时进行SpeI酶切后连接,并转化大肠杆菌感受态。利用菌落PCR对eGFP表达盒的连接方向进行鉴定,得到的阳性克隆送上海英骏生物技术有限公司测序。
测序结果返回后,利用DNASTAR软件与转移载体序列进行比对。经Oligo6.0软件分析,菌落PCR鉴定的引物对可以采用eGFP表达盒上游引物EU和同源重组臂右臂下游引物RL。eGFP表达盒正向插入时电泳结果为一条带大小为2 323 bp,即表达盒与右臂序列长度之和,反向插入时为两条不亮的条带且两条带的产物相差721 bp。转移载体构建示意图见图。

图1 转移质粒p UC18-LR-eGFP的示意图
选择lipofectaming 2000作为转染试剂,细胞转染密度为90%。转染在6孔细胞板上进行,细胞接种密度为3×105/ml。细胞生长对数期是转染的最好时机,通过培养基颜色变化和显微镜下细胞生长状态判断细胞生长的对数期。
各以250μl无血清无双抗的DMEM/F12分别稀释脂质体和质粒,在5 min内轻柔混合,配制不同比例的脂质体和质粒DNA混合物,室温静置孵育20 min;将脂质体和质粒混合物500μl/孔轻轻逐滴加到已含1.5 ml/孔培养液的6孔板中,轻轻十字形晃动使混合物均匀铺上LMH细胞,于5%CO2 37℃培养,转染5 h后更换为含5%血清、100 U/ml青链霉素的培养液继续培养,待转染24~48 h后观察瞬时转染情况。
FAdV-1 AV208*干粉冻**复液(对鸡胚肾细胞的TCID50为105.5/0.5 ml)用2%维持液1∶1 000稀释接种LMH细胞,感染2 h后弃病毒稀释液加入新鲜无血清无脂质体培养基1.5 ml/孔,脂质体和质粒转染细胞方法见1.7.2,每隔24 h观察eGFP表达及细胞病变情况。
当80%以上细胞出现明显CPE时收获细胞,每孔弃部分上清,余约1 ml细胞上清,用细胞刮将细胞刮下来收至1.5 ml Ep管,冻融一次4℃保存备用。
收集的单孔1 ml细胞培养物初次传代时接2块6孔板,每隔24 h观察eGFP的表达及细胞病变情况,在细胞病变较明显但不是特别严重时,在荧光显微镜下,用Marker笔圈取尽可能大的呈现绿色荧光斑的细胞团。
在生物安全柜中,用细胞刮和Tip头,将细胞团圈住部分之外的细胞尽可能的刮干净,并用培养液轻轻漂洗多次,然后在荧光显微镜下再次确认圈住的荧光斑细胞团附着在6孔板上,用适量培养液吹散混匀,收取至1.5 ml Ep管,冻融一次,取200μl收集液以1∶100稀释接种到一块6孔板上继续感染LMH细胞。

重复上述挑取荧光斑细胞团的步骤来筛选重组病毒。待多轮筛选后,病毒混合液中重组病毒的比例较大时,可将病毒混合液作倍比稀释稀接种LMH细胞,通过蚀斑技术纯化重组病毒。
将收获的病毒液以无血清DMEM/F12培养基10倍比稀释后感染LMH细胞,吸附1 h后弃去细胞表面液体,加入2 ml 2×DMEM/F12(含有0.02%中性红染液和4%血清)和2%低熔点琼脂糖等体积混合凝胶,待出现明显的蚀斑时,在荧光显微镜下观察是否有绿色荧光出现。
挑取有绿色荧光的蚀斑放入1 ml无血清的DMEM/F12培养液中,反复冻融3次后接种细胞进行新一轮的蚀斑纯化,直至所有病毒蚀斑在均呈现绿色荧光为止。将纯化到的重组病毒扩大培养。
在同源臂接近缺失片段的位置设计一对引物,序列如下:

用重组病毒rFAV-I-eGFP感染细胞,病变完全后收取细胞培养物,SDS-蛋白酶K法提取重组病毒的核酸DNA,用上述引物做PCR鉴定。若扩增的片断为1 600 bp的表达盒片段,而无3 000 bp左右的缺失片段,则表明已经纯化完全。
PCR获得了长度为4 111 bp和438 bp的基因组片段,经测序后获得的序列拼接后,与CELOV基因组进行序列比对后发现,此4 454 bp序列与NCBI数据库提供的CELOV基因组nt39 294~nt437 4 7之间同源性高,仅存在5个碱基缺失差异。
PCR扩增和测序结果均显示,得到了771 bp的左臂片段L和721 bp的右臂片段R。
中间载体pUC-L-R的测序结果返回后,利用DNASTAR软件进行比对分析,显示左右同源重组臂已通过共有的Spe I位点相连接。
经测序确认,引物对EU/EL扩增出了1 602 bp的完整的eGFP表达框。
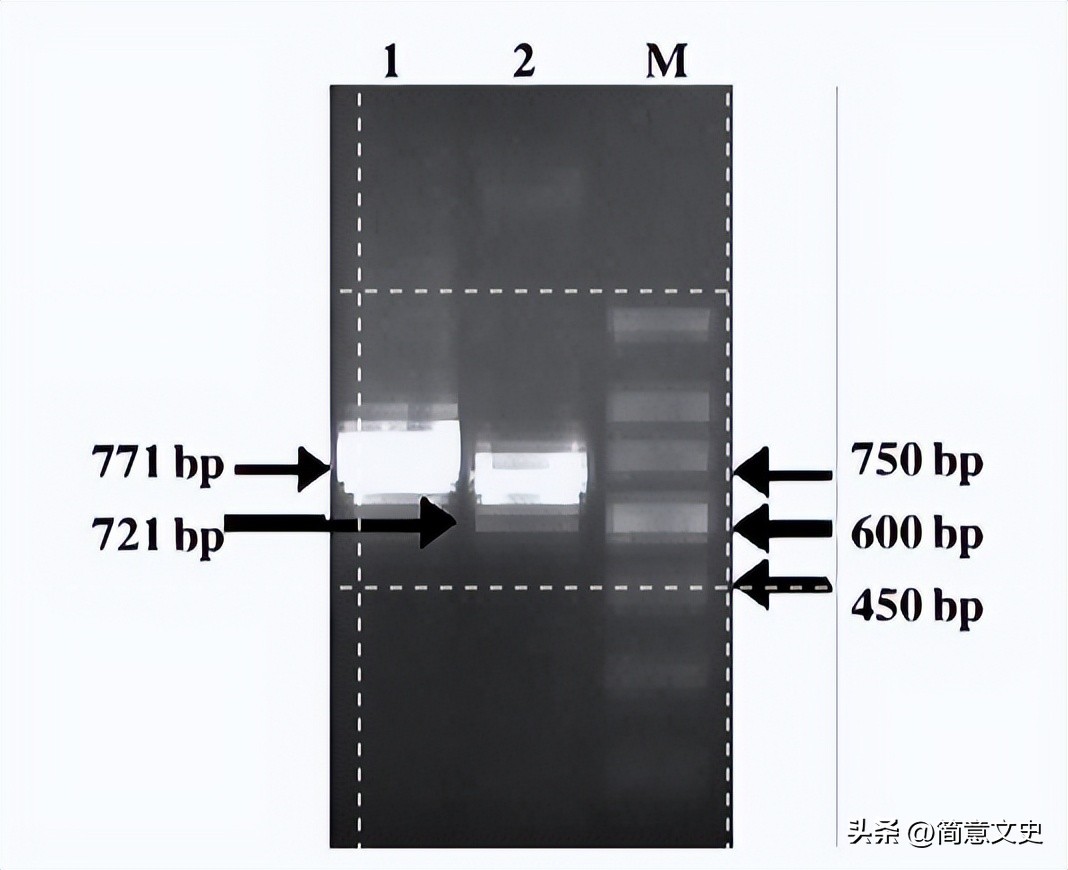
图2 721 bp、771 bp片段扩增结果
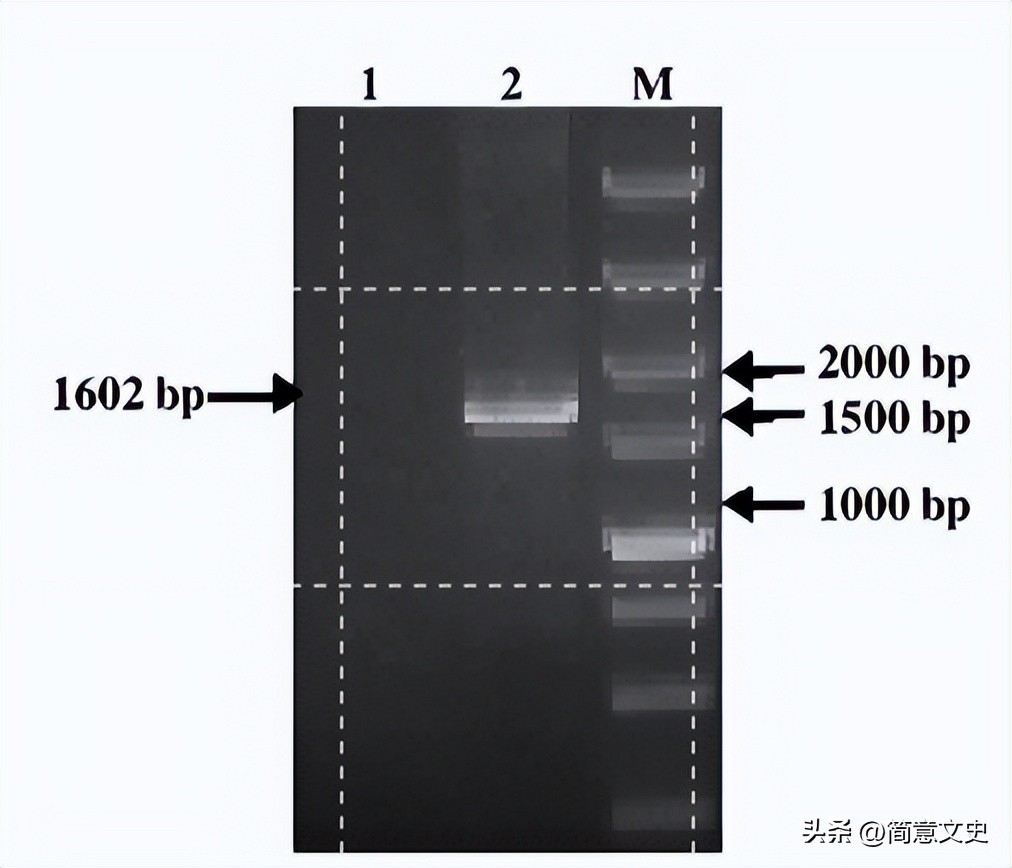
图3 1 602 bp片段扩增结果
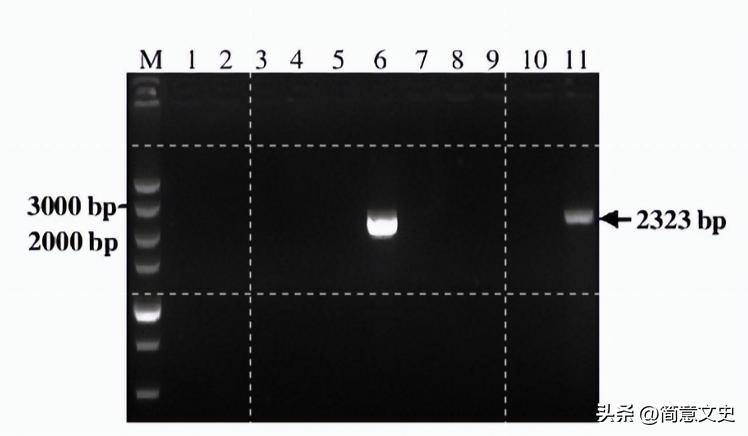
图4 菌落PCR产物电泳
利用菌落PCR对eGFP表达盒的连接方向进行鉴定,得到两个疑似阳性克隆,条带大小为2 323 bp。测序结果返回后,与DNASTAR软件拼接的左右同源重组臂及eGFP表达框共3 103 bp序列进行比对,结果完全一致。
最佳转染条件为细胞以3×105/ml密度接种于6孔板,36 h后细胞密度达到90%进行转染。
根据转染后细胞的荧光表达量结果,确定转移质粒pUC18-LR-e GFP与脂质体最佳转染剂量为用8μl lipofectamine2 000试剂和3μg pUC-LR-GFP质粒转染。
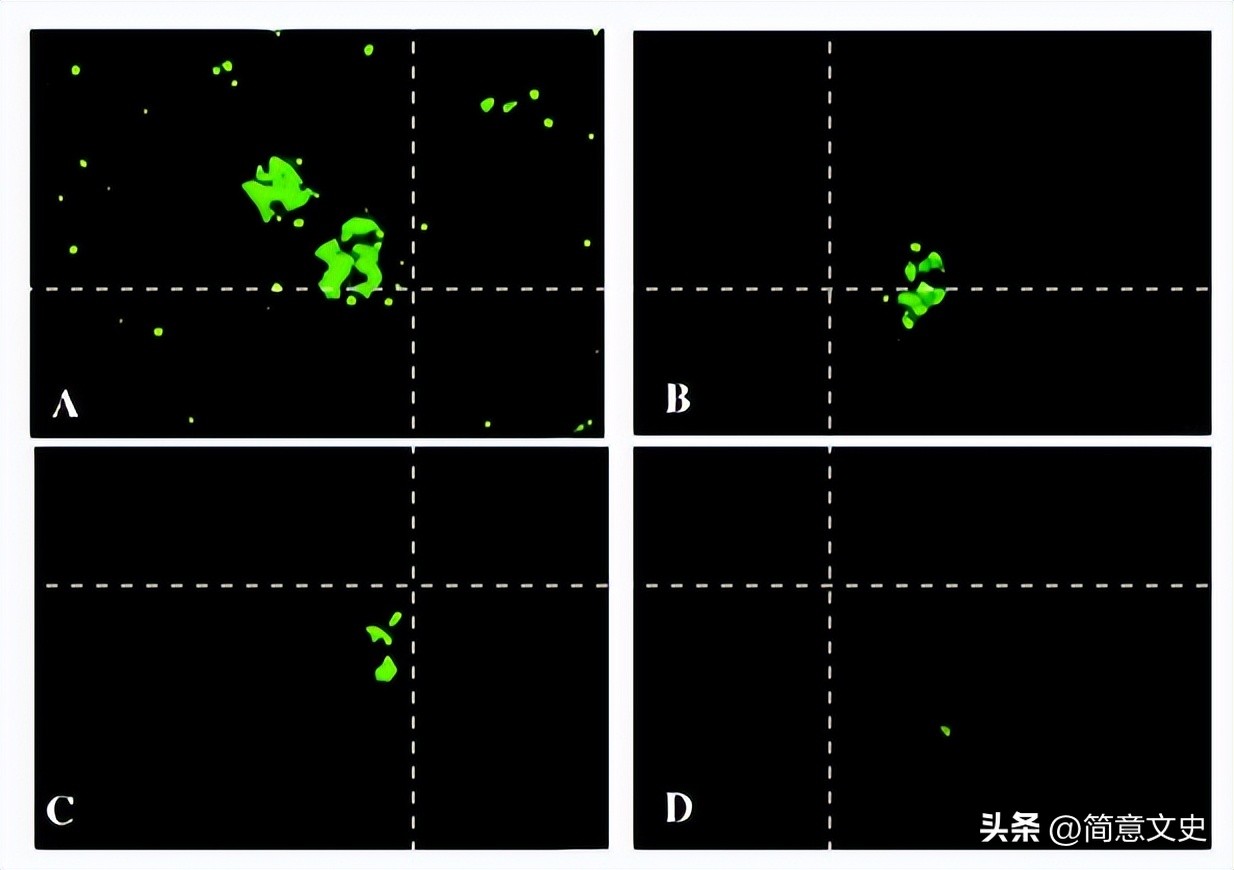
FAdV-1感染LMH细胞2 h后,转染重组质粒pUC-LR-eGFP,24~48 h在荧光显微镜下观察到较强的瞬时表达的eGFP;h后可看到明显的细胞病变。

收集的单孔1 ml细胞培养物初次传代时接2块6孔板,第3日即出现明显的细胞病变。挑取呈现荧光簇的细胞培养物在LMH细胞上传代第3次后,荧光镜下仍能观察到细胞走向的特异性荧光簇或散在荧光,说明转移质粒能够与FAdV-1在LMH细胞内发生同源重组,筛选到了重组病毒rFAdV-1-e G FP。
以病变的细胞培养物接种LMH细胞,用含1%低熔点琼脂糖的DMEM/F12培养细胞至出现明显的蚀斑时,挑取在在荧光显微镜下呈现绿色荧光的蚀斑进行下一轮的纯化,多次纯化后挑取蚀斑扩大培养后鉴定。
纯化至第4代,重组病毒PCR扩增结的片断为1 600 bp的表达盒片段,而无3 000 bp左右的缺失片段,表明已经纯化完全。
常用的重组腺病毒系统在载体构建操作复杂性和包装效率上存在差异,各有优缺点,很难有一种方法完全通用于各种重组腺病毒的构建。目前在临床上应用的腺病毒载体系统基本是第一代腺病毒系统,主要是通过细胞内同源重组的方法构建。
这种构建方法可行的最关键因素在于系统的稳定性,已有前人大量的研究工作证实。本研究通过选择新的亲本毒株和改进载体构建方法,期望将FAdV-1构建成更适合禽类疫苗研制的基因转移载体。
通过细胞内同源重组构建复制型重组病毒涉及多个方面,包括亲本毒株的选择、复制非必需区的确定、同源重组臂的大小、骨架质粒的选取、外源基因表达盒的设计等。
本研究选用FAd V-1构建重组禽腺病毒载体,参考了前人对CELOV复制非必需区的研究,CELOV右末端区域(nt40 065~nt43 684)是目前已鉴定的最大复制非必需区,利用这一区域构建CELOV基因转移载体,成功表达了许多外源蛋白。

构建用于重组的转移载体时,同源重组臂应达到一定长度,根据前人的研究结,同源重组臂长度在100 bp~1 000 bp长度就可发生同源重组。左右同源重组臂分别为771 bp和721 bp,保证了较高的同源重组效率。
转移载体p UC18全长仅2 686 bp,骨架质粒越小,将越容易转染。在外源基因表达盒的设计中,不仅插入了报告基因方便重组病毒的筛选,还引入了多克隆位点将转移载体构建成通用型载体,方便外源基因的插入。
尽管做了较大程度的缺失,重组禽腺病毒的包装容量仍是有限的,而源自质粒pe GFP-N1的包括e GFP基因、CMV启动子、MCS和poly A信号在内的各种表达元件加起来才1602 bp,可一并克隆至转移载体。
本研究中引入的MCS在用于外源基因插入时有四个酶切位点不可用,两个为同源重组臂与骨架载体的两末端接头Eco RI和H in dⅢ,两个为同源重组臂上左臂上的Kpn I和右臂上的Nhe I位点。
只有在e GFP表达框和缺失复制非必需区的腺病毒基因组上均不存在而在多克隆位点上存在的酶切位点才能成为单一酶切位点,用于外源基因插入。在非必需区的缺失策略上,作者在两同源重组臂靠近缺失区域的一端引入了相同的酶切位点,同源重组臂之间可通过共有的酶切位点相连接缺失掉中间的复制非必需区。
通过一步引入完整的外源基因表达盒和人为地添加适当的酶切位点,改进了载体构建方法,极大的减少了工作量。