免责声明:本人所有回答均参考自人民卫生出版社教科书,不构成任何医疗建议,只做医疗信息传播,如有身体不适请于正规医院就诊。
本人是神经内科主治医师,擅长脑血管病,帕金森病,神经脱髓鞘病变等领域,欢迎大家点击右上角关注:温州神经内科李晟医生,了解更多健康知识,请百家号抄袭我时可以表明出处:*今条头日**温州神经内科李晟医生思维导图,谢谢!
从今天开始每天奉上一张思维导图,一天一个医学知识分析
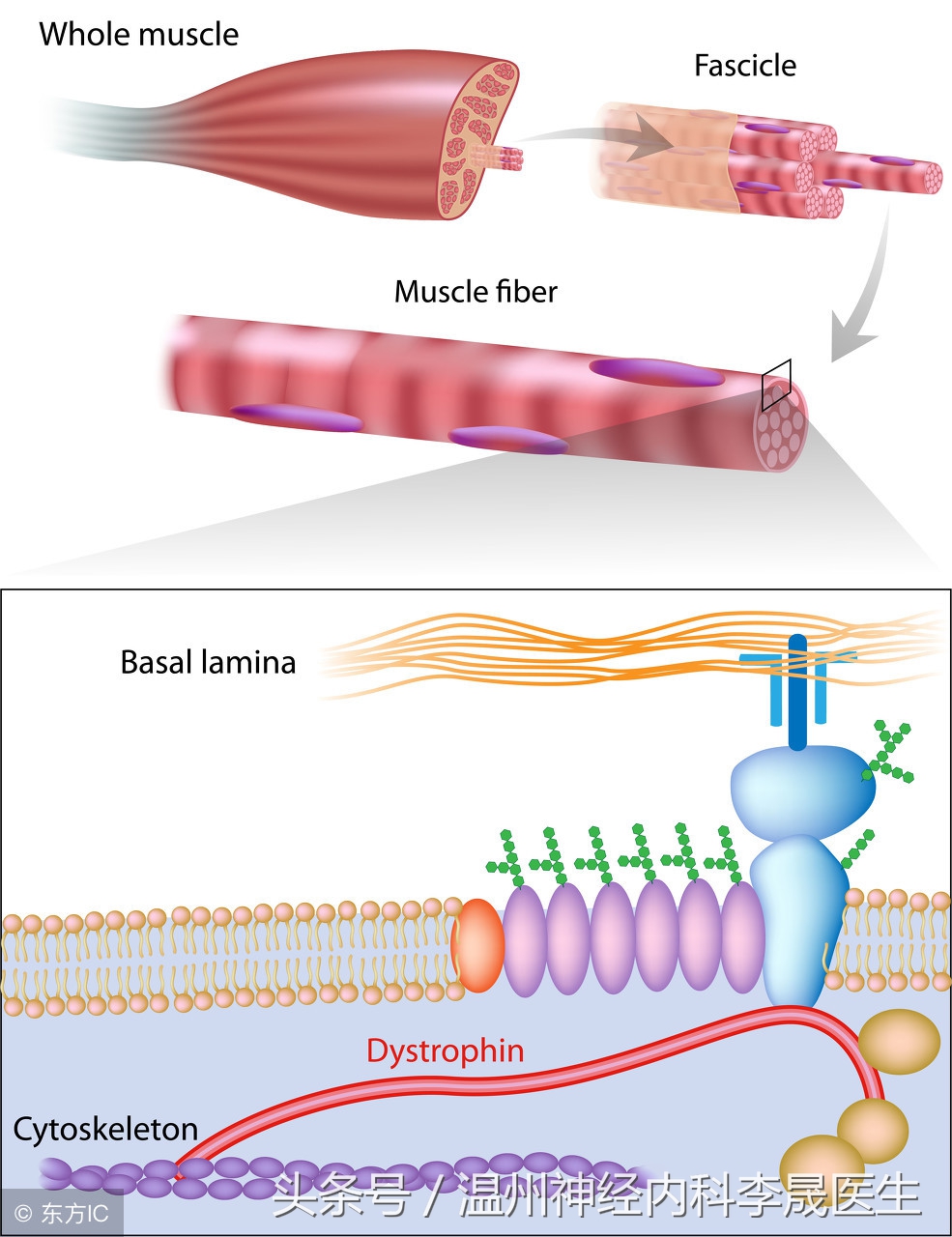
进行性肌营养不良症(progressive muscular dystrophy,PMD)
是一组遗传性肌肉变性疾病,临床特征主要为缓慢进行性加重的对称性肌肉无力和萎缩,无感觉障碍。遗传方式主要为常染色体显性、隐性和X连锁隐性遗传。电生理表现主要为肌源性损害、神经传导速度正常。组织学特征主要为进行性的肌纤维坏死、再生和脂肪及结缔组织增生,肌肉无异常代谢产物堆积。治疗方面主要为对症治疗,目前尚无有效的根治方法。
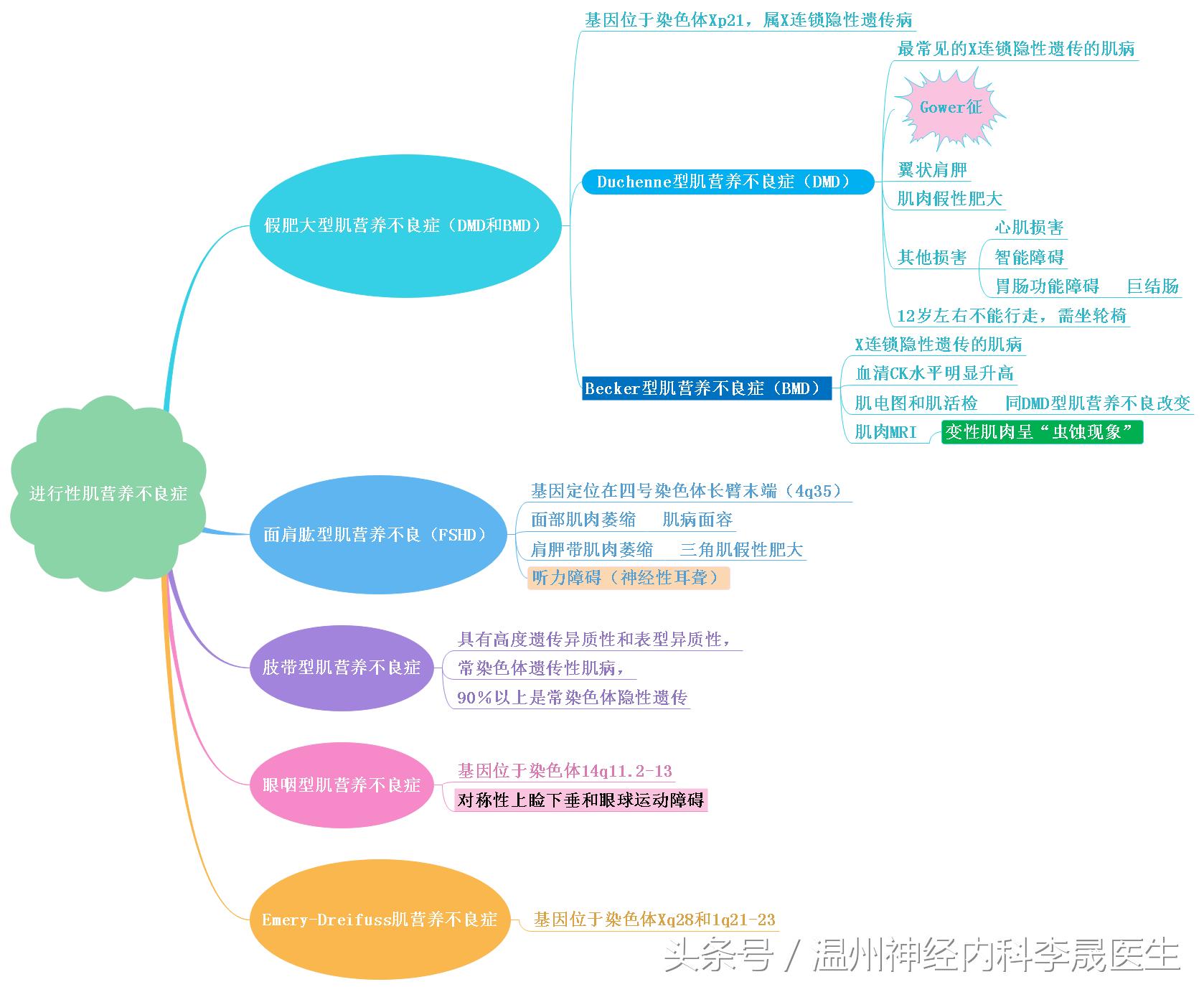
根据遗传方式、起病年龄、萎缩肌肉的分布、病程进展速度和预后,进行性肌营养不良症至少可以分为9种类型:
假肥大型肌营养不良症,包括Duchenne型肌营养不良症和Becker型肌营养不良症
面肩肱型肌营养不良症
肢带型肌营养不良症
Emery-Dreifuss肌营养不良症
先天性肌营养不良症
眼咽型肌营养不良症
眼型肌营养不良症
远端型肌营养不良症
-
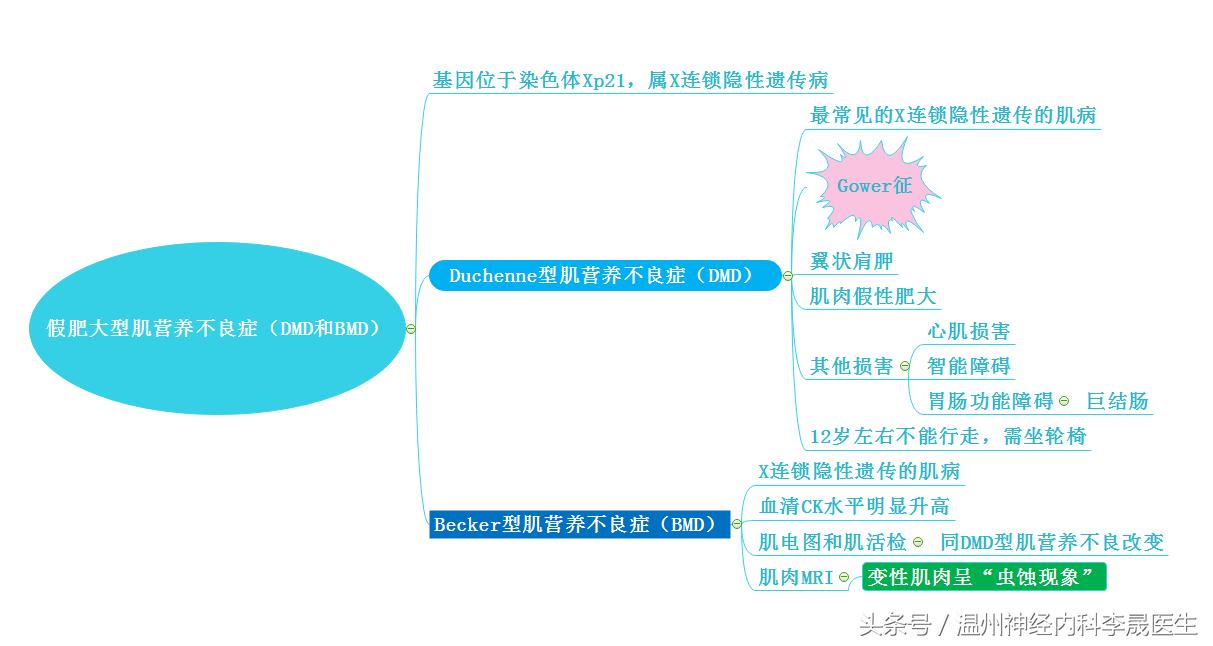
Duchenne型肌营养不良症(DMD)
1)DMD是我国最常见的X连锁隐性遗传的肌病.
2)Gower征, Gower征是DMD的特征性表现

Gower征
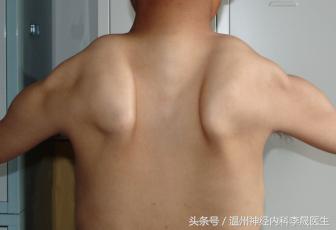
3)翼状肩胛
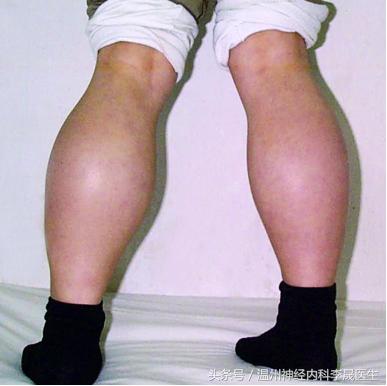
4) 肌肉假性肥大
5) 其他损害:
心肌损害(多数有):心律不齐,深Q波,心脏扩大,心瓣膜关闭不全
智能障碍;30%出现,程度不等
胃肠功能障碍:腹痛、吸收差、巨结肠
面肌、眼肌、吞咽肌、胸锁乳突肌和括约肌一般不受累及
2.Becker型肌营养不良症(BMD)
BMD和DMD一样,也呈X连锁隐性遗传
临床表现与DMD类似:累及骨盆带肌、下肢肌,有腓肠肌的假性肥大
血清CK水平明显升高,尿中肌酸增加,肌酐减少
肌电图和肌活检:同DMD型肌营养不良改变
肌肉MRI:变性肌肉呈“虫蚀现象”
3.面肩肱型肌营养不良症(FSHD)
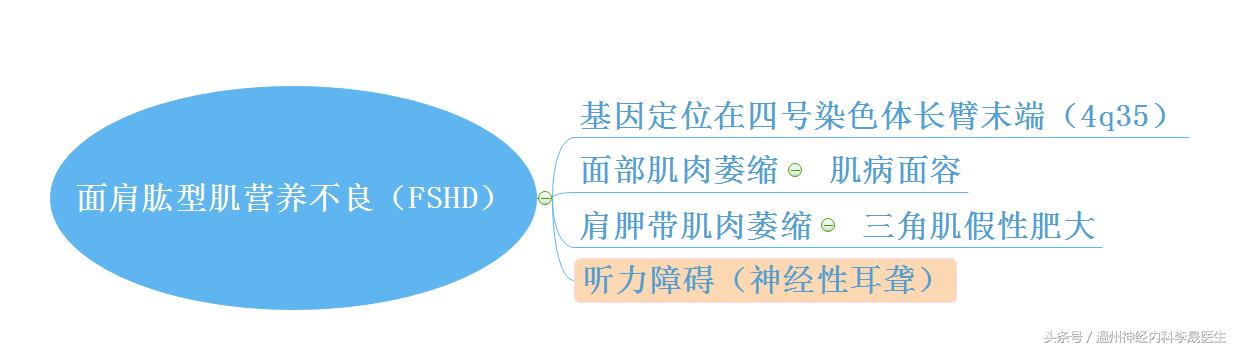
1).常染色体显性遗传,多见青少年期起病
2).面部和肩胛带肌肉最先受累
面部肌:面部表情少。肩胛带肌受累:翼状肩胛
口轮匝肌:假性肥大嘴唇增厚而微翘称“肌病面容”
三角肌假性肥大
3).病情进展缓慢,躯干和骨盆带肌肉,也有腓肠肌假性肥大,视网膜病变,听力障碍(神经性耳聋),20%需坐轮椅。生命年限接近正常
4).实验室:肌电图为肌源性损害,血清肌酶正常或轻度升高
5).确诊方法:印迹杂交DNA分析测定4号染色体长臂末端3.3kb/KpnI重复片段的多少
4.肢带型肌营养不良症
1)常染色体隐性或显性遗传,多为散发
2)隐性遗传常见、症状较重、起病较早
3)10-20岁起病,首发为骨盆带肌肉萎缩,逐渐出现腰椎前凸,鸭步,上楼困难,腓肠肌假性肥大,翼状肩胛。面肌受累少
4)病后20年丧失劳动力
5)肌电图肌源性损害,肌酶明显增高,心电图正常

5.眼咽型肌营养不良症
1.常染色体显性遗传
2.40岁左右起病
3.首发症状:对称性上睑下垂和眼球运动障碍。逐渐出现轻度面肌、眼肌无力和萎缩、吞咽困难、构音不清,近端肢体无力。易与MG混淆
4.血清CK正常或轻度升高

6.Emery-Dreifuss型肌营养不良症
1.X连锁隐性遗传,5~15岁缓慢起病
2.临床特征:肘部屈曲挛缩和跟腱缩短,颈部前屈受限,脊柱强直而弯腰、转身困难
3.心脏传导功能障碍:如心动过缓、晕厥、心房纤颤,心脏扩大,心肌损害明显。常因心脏病而致死
4.血清CK轻度增高。病情进展缓慢

特鲁多铭言——To Cure Sometimes, To Relieve Often, To Comfort Always.(有时治愈,常常帮助 ,总是安慰 )致医生,致患者,请记住医生与患者最大的敌人永远是疾病。
