
医脉通编译,未经授权,请勿转载。
病史描述
杰斯·L·卡普兰医师(儿科):一例9月龄的男孩因发热、腹泻和肝功能衰竭而入院。
患儿在入院前2天情况良好。入院当天出现嗜睡、易激惹、鼻漏、间歇性呕吐和非血性腹泻,伴经口摄入(量)减少。次日,据报告,体温升至38.1°C。患儿早期就诊于一家与本院有关系的卫生所;当时体温为37.6°C,体格检查正常。患儿拟诊为病毒性胃肠炎,(医生)给予对乙酰氨基酚和小儿电解质口服液,院外治疗。次日,症状加重,夜间就诊于本院急诊科。
患儿足月,阴道分娩;(出生后)1分钟和5分钟的阿普加评分分别为8分和9分。常规新生儿代谢筛查的结果正常。患儿在出院前接受了乙型肝炎疫苗(接种)。据报告在(本次)入院前1个月,他曾有一次呼吸道感染;在入院前3天,他首次迈步时曾经摔倒,并且擦伤了其前额。他的其他方面都好,生长发育正常。他仅有的用药是1天的对乙酰氨基酚(120 mg,根据需要每4~ 6小时使用1次治疗发热);据报告,目前为止,该患儿及时接受了儿童期的免疫接种。他没有任何已知的变态反应。他与其母亲住在一起,其母亲曾有呼吸道症状达1周,并接受了阿奇霉素治疗。他未被送至日托所,也没有已知的旅行或毒物暴露(包括摄入蘑菇)。他的母亲在童年期曾有抽搐史。
体格检查,患儿倦怠,间歇性不安,并且啜泣。体重是8.9 kg(相应年龄的第25至第50百分位数),体温37.6°C,血压95/46 mgHg,脉率133次/分;呼吸率26次/分,以及在他呼吸周围空气时的氧饱和度为100%。结膜苍白、无黄疸。左眼上方有一块瘀伤。肠鸣音减弱,腹软,无压痛或膨隆。肝边缘延伸至肋缘下4 ~ 5 cm。其余检查均正常。镁、C反应蛋白、淀粉酶、脂酶、铁、总铁结合力和乳酸盐水平均正常;其他结果显示于表1。尿液分析正常,尿液毒物筛查阴性。腹部X线检查显示非特异性肠内积气征,没有游离气体或肠梗阻的证据。给予静脉液体输入。没有使用对比剂的脑部计算机体层摄影(CT)检查正常。腹部的超声检查显示肝脏回声略微增强,并且胆囊周围有液体,没有肝内或肝外胆管扩张的证据。
在采用咪达唑仑镇静后,使用口服和静脉注射对比剂的胸部、腹部和盆腔CT显示,肝脏显著增大,并有符合脂肪浸润的弥漫性低衰减(信号)和胆囊周围液体。CT检查在其他方面正常。
患儿被收入儿科重症监护病房。行经股静脉中心静脉置管术,持续监测心血管和呼吸变量,限制肠内营养。给予N-乙酰半胱氨酸和头孢曲松,使用雷尼替丁和维生素K,并给予劳拉西泮和*啡吗**治疗躁动。血清对乙酰氨基酚的水平<10.0 mg/L(参考范围10~25 mg/L),(以上药物)在尿检中未被检出。在前2天中,血清异嗜性抗体检测和爱泼斯坦-巴尔(Epstein-Barr)病毒(EBV)、甲型和丙型肝炎病毒检测为阴性,以及在一份鼻样本中进行的呼吸道病毒(腺病毒、甲型和乙型流感病毒、副流感病毒以及呼吸道合胞病毒抗原)检测也为阴性;乙型肝炎病毒表面抗体的检测为阳性,表面抗原为阴性。血、尿和便培养均无菌(生长)。其他实验室检查的结果显示于表1。
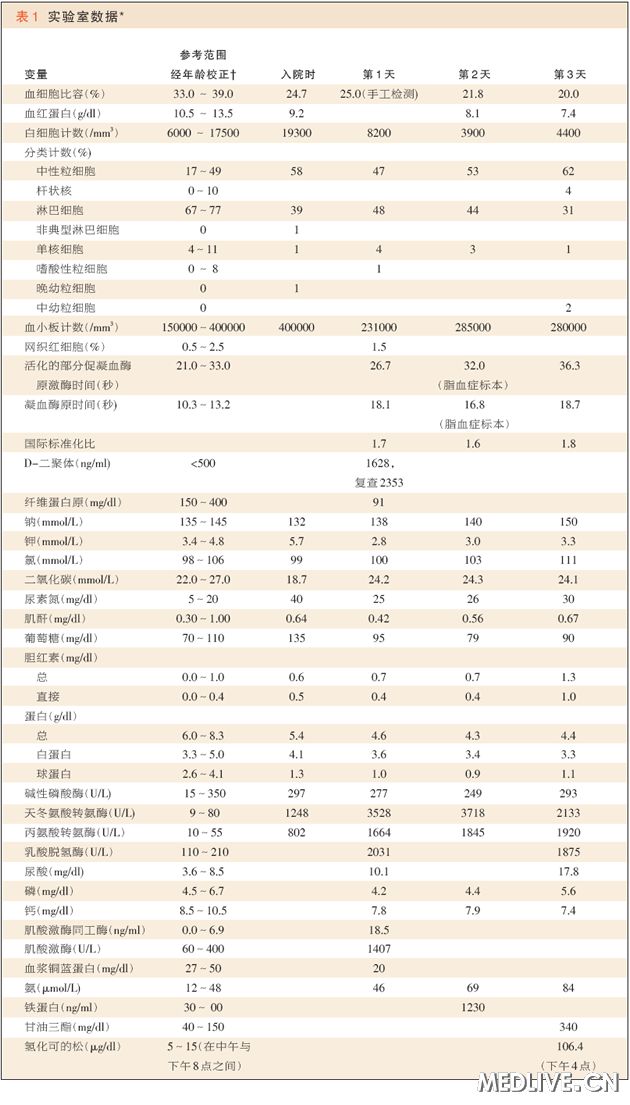
表1 实验室检查(* 单位换算:尿素氮mmol/L=mg/dl×0.357,肌酐μmol/L=mg/dl×88.4,葡萄糖mmol/L=mg/dl×0.05551,磷mmol/L=mg/dl×.3229,钙mmol/L=mg/dl×0.250。† 参考值受许多变量影响,包括病人人群和实验室使用的方法。马萨诸塞总医院使用的范围是经年龄校正的,并且针对未妊娠和未患可影响结果的内科疾病的病人。因此,这些范围不一定不适合于所有病人。)
在接下来的2天中,(患儿)发生了低血压和低氧血症,收缩压60~70 mmHg,舒张压30~40 mmHg,以及当患儿呼吸周围空气时的氧饱和度为88%;嗜睡加重,伴瞳孔反应迟钝。给予鼻导管吸氧和推注生理盐水;血压恢复正常但随后下降;给予多巴胺和去甲肾上腺素。心电图显示窦性心律,(心率)153次/分,伴不完全右束支传导阻滞和右室肥大;超声心动图检查正常。头部CT检查正常。
梅毒、弓形虫抗体和细小病毒B19抗体、巴尔通体(免疫球蛋白IgG和IgM)、水痘-带状疱疹病毒(IgG)以及巨细胞病毒(CMV)抗原检测均为阴性;其他结果显示于表1。在第3天,(患儿)输入了红细胞;血细胞比容增加至31.4%。肝位于肋缘下6~8 cm,延伸至脐部并且触诊坚硬。其他检测结果有待回报。
影像学讨论
玛莎·凯·费林医师:我们可以回顾一下影像学检查吗?
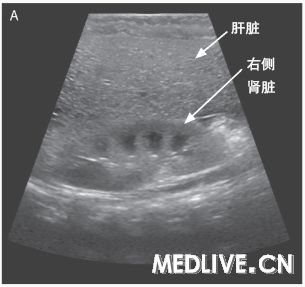

图1 腹部成像检查 右肝叶矢状面超声图像(图A)显示,与邻近的肾脏相比,肝实质回声增强,这与脂肪浸润(的表现)一致。腹部轴向计算机体层摄影图像(图B)显示,弥漫性肝脏密度降低,这也与脂肪浸润(的表现)一致。
奥托·拉帕利诺医师:入院时进行的腹部超声显示肝实质回声弥漫性增强,符合脂肪浸润,没有局灶异常,并且胆囊周围有少量液体(图1A)。为了进一步评估肝脏而进行的腹部CT检查显示,肝实质脂肪性衰减,轻度肝肿大,以及胆囊周围有少量液体(图1B)。
埃斯特·J·伊斯雷尔医师:我知道这个病例的诊断。由于担心急性肝衰竭,我们请小儿胃肠病和肝病科(医师)看了这例婴儿。儿童急性肝衰竭的(诊断)标准包括在预先不存在肝病的情况下转氨酶水平升高≤8周,国际标准化比(INR)>1.5(或凝血酶原时间≥15秒)并且有脑病,或INR>2.0(或凝血酶原时间>20秒)但没有脑病1。在第3天,我们(这例)患儿的INR为1.8,凝血酶原时间为18.7秒,并且在第4天INR增至>2,因此他符合肝衰竭的标准。难以确定他是否有脑病,因为他在第1天因置管而接受了咪达唑仑和*啡吗**治疗,并且此后他昏睡,至少2天对疼痛没有反应。
(我们)开始给予(患儿)预防性抗生素(治疗),并且加用N-乙酰半胱氨酸,尽管该药在儿童非对乙酰氨基酚相关急性肝衰竭中的疗效尚不确定。该婴儿还接受了肝移植可能性的评估。急性肝衰竭的原因随后被评估。
在这例患儿中,肝衰竭的鉴别诊断包括对乙酰氨基酚和其他药物的使用[这是最常被识别的肝衰竭的原因(16%的病例)]、代谢障碍(10%)、病毒(6%)和缺血(4%)2。儿科急性肝衰竭数据库显示,在儿童患者中,49%肝衰竭原因不明,尽管这种情况因儿童的年龄而异2。这例患儿的年龄为9个月;在婴儿中,代谢障碍(性肝衰竭)比药物相关原因的肝衰竭更常见。在一项对文献的回顾性研究中3,42%的急性肝衰竭婴儿涉及代谢性原因,并且在该年龄组中仅16%的病例原因未确定。儿科急性肝衰竭数据库最近的一项评价显示,原因不明的急性肝衰竭的患儿并未得到代谢性和自身免疫性原因的全面评估4。
在这一病例中,可能的感染性原因包括单纯疱疹病毒、细小病毒、人疱疹病毒6型、CMV、EBV以及甲型、乙型和丙型肝炎病毒(感染);这些病毒的检测均为阴性,便、尿和血的培养也均为阴性。此外,大多数由原发性肝脏感染所致肝衰竭的病人表现为高胆红素血症和黄疸,而我们这例患儿胆红素水平正常。
急性肝衰竭有可能是自身免疫性肝炎的最初表现,尽管这些疾病较常表现为慢性疾病。应进行1型自身免疫性肝炎H的抗核抗体和抗平滑肌抗体以及2型(自身免疫性肝炎H)的抗-肝-肾微粒体抗体的评估。尽管2型自身免疫性肝炎可见于幼儿,但它极少见于婴儿。
这例婴儿接受过对乙酰氨基酚(治疗),据报告为正常剂量,并且他有低的血清对乙酰氨基酚水平,但是否存在一次意外过量也应该考虑。药物和毒素所致的肝损伤可以是剂量依赖性或特应性的。儿童期肝衰竭病例所涉及的毒物包括条蕈(毒蘑菇)和有机溶剂(例如四氯化碳和草药)5。此患儿没有暴露于任何这些毒素,并且毒素筛查为阴性。
我们考虑了由肝静脉闭塞引起的缺血性疾病[巴德-恰里(Budd-Chiari)综合征(肝静脉反流障碍综合征)],但除了肝大和腹水之外,该病通常还伴有极显著的高胆红素血症。CT显示肝静脉通畅。在该年龄组中,急性肝衰竭作为噬血细胞淋巴组织细胞增生症的一种表现也必须予以考虑;这例患儿有升高的铁蛋白和甘油三酯水平以及低纤维蛋白原血症,但他没有发热、脾大或影响1个以上细胞系的血细胞减少,使这一诊断不太可能。
排除这些诊断后,给我们剩下了代谢性疾病。在婴儿早期出现的代谢性疾病通常伴有胆汁郁积,而这例婴儿没有(胆汁郁积)。在这例年龄较大的婴儿中,脂肪酸氧化缺陷、胆固醇酯贮积病、糖原贮积病和囊性纤维化相关的肝病必须予以考虑6。威尔逊(Wilson)病(肝豆状核变性),是年龄较大的儿童和成人中可表现为急性肝衰竭的最常见代谢性疾病,在3岁以下儿童中尚未被报告。这例患儿的年龄,肝脏成像检测出存在脂肪,以及在住院的最初几天中肝脏大小迅速增加,这使我们同时考虑了脂肪酸氧化缺陷以及胆固醇酯贮积病。尽管他有凝血障碍,但我们还是决定进行一次肝活检以明确诊断。
病理讨论
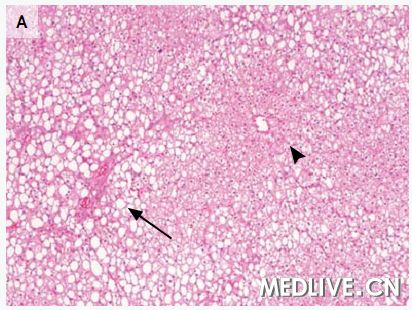
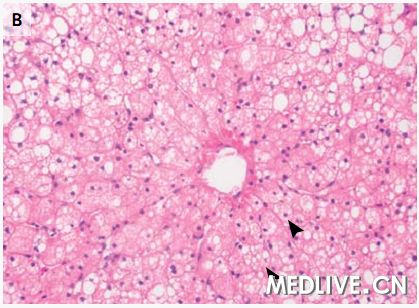

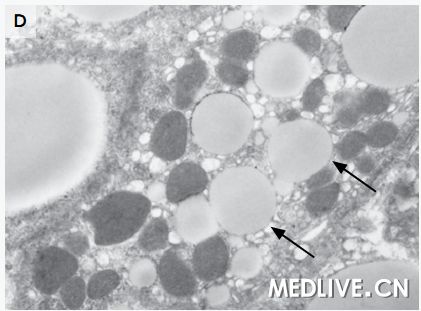
图2 肝活检标本 在低倍放大时(图A),一种小泡型的脂肪变性主要存在于(肝)中央小叶区(箭头),以及大泡型存在于门管区周围(箭)。在较高倍放大时(图B),广泛性小泡性脂肪变性存在于中心小静脉周围肝脏的中央小叶区(箭头)。被膜下区(图C)显示广泛的小泡性脂肪变性(箭头)。超微结构检查(图D)显示一个肝细胞含有不明显的线粒体和非膜结合性脂质囊泡(箭)。
米哈伊尔·利索夫斯基医师:(我们)进行了一次肝脏楔形活检。活检标本显示弥漫性重度脂肪变性(图2A、2B和2C)。小泡性(脂肪变性)形式主要见于小叶中央区,而大泡性(脂肪变性)形式存在于门管周围区域。没有肝炎、肝细胞坏死、纤维化或胆小管反应的证据,并且没有巨线粒体或可极化物质。使用淀粉酶消化的过碘酸-希夫(Schiff)染色显示肝细胞中没有胞浆内小体,并且没有糖原蓄积。铜和铁的染色以及黏多糖的胶体铁染色为阴性。电子显微镜观察证实肝细胞中存在丰富的非膜结合性脂质囊泡(图2D),并且没有显示线粒体异常、糖原沉积或其他的蓄积产物。(我们)做出了弥漫性小泡性和大泡性脂肪变性的诊断。
小泡性脂肪变性由线粒体功能受损导致,在这种情况下,脂肪酸β氧化减少导致三酰甘油(triacylglycerol)和游离脂肪酸的蓄积7。在本例患儿的年龄组中,线粒体功能障碍通常由一种遗传性代谢错误或一种药物或毒素的作用所致8。遗传缺陷可涉及线粒体的氧化磷酸化、脂肪酸氧化或尿素生成系统。门管周围糖原化的核(glycogenated nuclei)、门管纤维化和肝细胞糖原聚集物的缺乏并不支持尿素生成障碍9。与线粒体氧化磷酸化异常相关的胆小管反应和纤维化并未被观察到10。我们考虑到了沃尔曼(Wolman)病(由溶酶体酸性脂酶无活性引起)以及胆固醇酯贮积病(由溶酶体酸性脂酶活性降低引起),但缺乏泡沫样巨噬细胞和库普弗细胞、可极化胆固醇酯晶体、纤维化,以及电子显微镜下没有膜结合性脂质和晶体并不符合这种诊断11。
小泡性脂肪变性可由一种药物或毒物[例如阿司匹林(Reye综合征)]、丙戊酸、核苷类似物(例如去羟肌苷和齐多夫定)、甲烯二氧甲苯丙胺、四环素、蜡样芽孢杆菌催吐毒素、急性铁超负荷或中毒导致。Reye综合征(脑病-脂肪肝综合征)在较大年龄组(4~15岁)中表现为全小叶型小泡性脂肪变性。(本例患儿)不存在蜡样芽孢杆菌毒性的肝细胞坏死特征12。急性铁中毒通过铁染色阴性而被排除13。其他药物原因根据临床情况已被排除。
因此,先天性脂肪酸β氧化缺陷仍然是主要的考虑。
鉴别诊断
费林医师:在一名9个月大的婴儿中,由脂肪肝的这种临床表现所提示的最有可能的先天性代谢缺陷显示于表2。
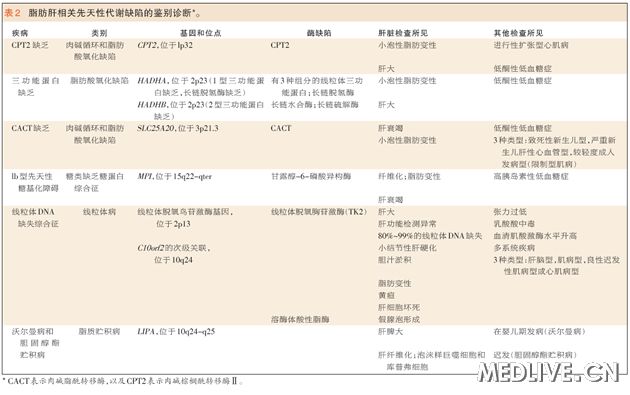
表2 脂肪肝相关先天性代谢缺陷的鉴别诊断
在一例有低酮性低血糖症的病人或一例有急性脂肪肝的婴儿(例如本病例)中,脂肪酸氧化缺陷和肉碱转运缺陷通常要被怀疑。有未经治疗的脂肪酸氧化缺陷的婴儿和(这种缺陷)肯定携带者的母亲(其发生妊娠脂肪肝的危险增加15),可发生脂肪肝。线粒体β氧化对于脂肪产生能量至关重要。长链脂肪由肉碱(β-羟基-γ-三甲胺丁酸)护运至线粒体内基质16。穿过线粒体外膜的主动转运由肉碱棕榈酰转移酶Ⅰ(CPT1)介导,而从外膜穿越至内膜由肉碱脂酰转移酶(CACT)介导,穿越线粒体内膜(的主动转运)由肉碱棕榈酰转移酶Ⅱ(CPT2)介导。在线粒体内,每“轮”循环都有2个碳[乙酰辅酶A(CoA)]从较长的链上被剪切下来,直到长链脂肪被还原为最后一个乙酰CoA。乙酰CoA被用于生酮作用[其对空腹期间的能量(产生)至关重要]和三羧酸循环[其被用于(产生)所有时间的能量,尤其是心脏和骨骼肌(的能量)]。涉及这一途径的任何酶的缺陷都可导致未氧化脂肪酸的蓄积,进而导致细胞内脂肪蓄积和各种器官功能障碍。
这例患儿直到9个月龄时一直很好,就在此时,(他)在有胃肠道疾病的情况下突然发生了急性肝衰竭。导致长链脂肪酸蓄积的脂肪酸氧化缺陷与短链和中链脂肪酸氧化缺陷相比,会表现出较严重的临床症状(低酮性低血糖症、肝病、骨骼肌病伴或不伴心肌病、猝死以及妊娠并发症)。后者发病较轻微,包括空腹失代偿(一种变异较大的低酮性低血糖症)和骨骼肌病。尽管如此,在婴儿期即使中链缺乏也可能是致死性的17,18。一些病人没有症状,直到一种疾病对机体代谢处理能力造成应激19,我们的患儿可能就是这种情况。考虑到这例患儿疾病的严重程度,我预测(患儿)存在一种会导致极长链脂肪酸蓄积的缺陷(如CACT或CPT2缺乏)。
在这例先天性代谢缺陷的可疑病例中,阐明诊断有赖于“生化指纹”—— 由于酶的缺乏导致一种特殊生物标志物的水平升高。在这一病例中,对经父母知情同意后获得的已经入档的原始新生儿筛查谱以及一份新鲜的血浆样本平行进行了检测。
玛莎·凯·费林医师的诊断
脂肪酸氧化缺陷(CACT或CPT2缺乏)。
讨论

图3 脑部MRI 和磁共振波谱分析 患儿的MRI(图A)与健康的9月龄对照者的MRI(图B)相比,T2-加权影像显示,在前叶白质和顶叶白质中有较广泛的信号减弱区,提示这些区域有成熟髓鞘形成。患儿的磁共振波谱分析(图C)显示了在大约1.4 ppm和0.9 ppm处的异常脂质峰(箭)。这些峰未见于健康对照者的磁共振波谱分析中(图D)。在患儿年龄为15个月时进行的磁共振波谱分析中,异常峰不再可见(图E)。
费林医师:取回的新生儿筛查谱分析显示,乙酰肉碱的中间产物(包括C18:1/C18:1OH、C16/C16OH和C14)水平升高,以及游离肉碱(C0)水平低。这种模式表明CPT2或CACT缺乏的可能性大。血浆样本确证了新生儿筛查的结果。这些升高包括C16水平升高至2.6 nmol/ml(参考范围<0.5 nmol/ml)和C18水平升高至0.59 nmol/ml(参考范围<0.12 nmol/ml)。血浆总肉碱水平为18 μmol/L(参考范围是38~68 μmol/L),以及游离肉碱水平为2 μmol/L(参考范围是27~49 μmol/L)。其他线粒体丢失检查显示平均线粒体DNA含量为41%(在正常范围内),不太可能是一种核编码的线粒体缺失缺陷。持续异常的肉碱水平强烈提示长链脂肪酸氧化途径和肉碱循环中的一种缺陷。
拉帕利诺医师:由于病人的精神状态发生改变,在住院第5天进行了一次3-特(斯拉)脑磁共振成像(MRI)检查及磁共振波谱分析。传统的T2-加权成像显示,与年龄相匹配的对照婴儿相比,(患儿)大脑两个半球的白质有明显的“成熟” 髓鞘形成(图3A和3B),没有局灶异常。从深部脑白质获得的单-体素磁共振波谱分析显示,与对照相比(图3D),在1.4 ppm和0.9 ppm处有明显的、界限清楚的异常脂质峰(图3C),以及N-乙酰天冬氨酸/肌酸以及胆碱/肌酸的比相对正常。
磁共振波谱分析中这些异常峰的放射学鉴别诊断包括数种脂质代谢障碍,包括施约格伦·拉尔森(Sjögren-Larsson)综合征(由微粒体脂肪醛脱氢酶缺乏导致)20-24、泽尔维格(Zellweger)综合征谱群疾病(PEX基因突变导致)25-27和CPT1缺乏28。
这例患儿没有施约格伦·拉尔森综合征的体征(鱼鳞病、精神损害和痉挛状态23,29),并且传统MRI检查没有脑室周围白质异常或髓鞘形成延迟的证据23,21,22。MRI检查结果也不支持泽尔维格综合征谱群疾病,该病在传统MRI检查中,通常伴有髓鞘形成延迟、皮质脑回形式异常和丘脑尾凹囊肿的证据27,29-31。文献已经报告了一例临床表现和磁共振波谱检查结果与本患儿相似的CPT1缺乏病例28。
费林医师:需要其他检查来证实诊断。可选择的(检查)包括对皮肤成纤维细胞或肌肉进行功能酶分析。然而,这些都是很费时间的检查,并且有赖于充分的成纤维细胞生长,来自皮肤活检样本的成纤维细胞生长可花费长达3周的时间。完成检查本身的时间范围可以从3周到8周32。
因为根据生化分析物鉴别诊断(的范围)已被缩小至CACT缺乏和CPT2缺乏,我们选择在贝勒医学院医学遗传学实验室李俊·C·王(Lee-Jun C. Wong)博士的实验室对这2种疾病的基因进行测序。CACT(SLC25A20)基因测序分析显示,在8号外显子中有一种F209X(804delG)突变(一种既往报告的致病性突变33);在2号外显子上有一种160G→T(G54W)突变,这种突变通过计算机模型被预测是一种假定的致病性错义突变;在2号外显子上还有一种163A→G(T55A)突变,这种突变根据计算机模型被推测是一种良性变异体。在CPT2 基因中没有检测到突变。
这一结果确立了CACT缺乏的诊断,该病是由SLC25A20基因的两个拷贝都发生突变而导致的常染色体隐性遗传性疾病(正如在本例患儿中所见的一样),导致CACT酶缺乏。其结果是,肉碱和乙酰肉碱穿越线粒体膜的交换被中断。这一年龄组(病人)的临床病程通常是严重的,伴有新生儿抽搐、心律失常和呼吸暂停。全球有不到30例的确诊病例34,35。
因德尼尔·萨海(Inderneel Sahai)医师:这例患儿有正常的新生儿筛查谱,并且当他的年龄为9个月时,一种先天性代谢缺陷被检出。在无症状的新生儿中,通过一种乙酰肉碱水平升高的特有模式可识别出脂肪酸氧化缺陷(包括CACT和CPT2缺乏),因此,它们可通过新生儿筛查被检出。然而,被纳入筛查谱的疾病依照儿童出生时所在州各自的现行法规来确定,州与州之间的法规存在极大差异。为了使全国的筛查标准化,美国医学遗传学学会建议普遍筛查29种核心疾病。另外25种次要疾病被列出,学会提出可报告(这些疾病的)检测结果36。这些次要疾病是那些在筛查核心疾病的过程中可以很容易地被识别,但其临床意义或合适的治疗尚不确定的疾病,因此,根据世界卫生组织(WHO)采用的威尔逊·琼格纳(Wilson-Jungner)标准,它们并不是筛查的理想候选疾病37。
尽管新生儿筛查在美国的大多数州都强制进行,但多数州允许父母决定不进行这种筛查38。马萨诸塞州强制对符合WHO标准的疾病进行筛查,并且对其他疾病提供可选择的筛查(需要父母知情同意),包括乙酰肉碱分析39。这例患儿的母亲拒绝了可选择的扩展检测项目。
这个病例强调了对临床上疑诊的代谢障碍(其通过筛查可检出)进行追踪的重要性,尽管新生儿筛查试验的结果正常亦如此。虽然假阴性筛查结果极为罕见,但仍可出现,因为分析物可由于下列情况而不存在异常:分析物浓度随时间发生变化,采集标本时的治疗干预,在婴儿室发生标本弄混或实验室差错。在这个病例中,如果我们没有仔细审查报告以确定其母亲拒绝了可选择的(筛查)项目,则正常的结果可能会引导我们考虑沃尔曼病而不是脂肪酸氧化缺陷。
治疗讨论
费林医师:大约75%的肉碱是从肉类、乳制品和大豆来源的饮食中获得的,25%通过内源性合成而生成。这例患儿接受去除饮食中的长链脂肪以及在其饮食中补充口服肉碱治疗。消化科还加用了熊去氧胆酸以减少脂肪的吸收。我们的长期策略将由一种多学科综合小组的方法组成,同时协调保健门诊、针对其酶缺乏的饮食和生化遗传学监督。避免空腹、多餐以及限制长链脂肪酸的特殊饮食是(治疗的)关键。
拉帕利诺医师:在患儿15个月大时,复查磁共振波谱分析显示,在诊断时看到的异常脂质峰已经消失(图3E)。
费林医师:患儿现在已经将近3岁,正在茁壮成长。他在膳食限制长链脂肪酸方面做得很好,开始采用一种婴儿元素配方,并且进展至含10%的必需脂肪酸、肉碱添加剂和熊去氧胆酸的食物。他的身高和体重位于第50百分位数,以及头围位于第35百分位数。所有发育过程中的指标患儿均已经达标。
南希·李·哈里斯医师(病理科):还有任何问题吗?
罗纳德·E·克莱曼医师(儿科):这个病例的许多意外检查所见中的一项是MRI上明显增强的髓鞘形成。是什么导致了这种情况?
拉帕利诺医师:(导致)这一检查所见的原因尚不清楚。可能的情况是,异常脂质代谢产物的存在可能引起这例患儿深部白质和皮质下白质明显的广泛性双侧“成熟”髓鞘形成。已有研究报告,斯特奇·韦伯(Sturge-Weber)综合征(脑面血管瘤病)40和半侧巨脑畸形41病人有单侧成熟髓鞘形成的区域,或在婴儿猝死综合征病例中有更广泛的报告42。然而,还缺乏关于脂质代谢障碍病人深部白质和皮质下白质(存在)明显的双侧成熟髓鞘形成的数据。
解剖学诊断
由CACT缺乏导致的小泡性肝脂肪变性。

参考文献:1. Squires RH Jr, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J Pediatr 2006; 148:652-8.
2. Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401-15.
3. Cochran JB, Losek JD. Acute liver failure in children. Pediatr Emerg Care 2007; 23:129-35.
4. Narkewicz MR, Dell Olio D, Karpen SJ, et al. Pattern of diagnostic evaluation for the causes of pediatric acute liver failure: an opportunity for quality improvement. J Pediatr 2009;155(6):801.e1-806.e1.
5. Campo JV, McNabb J, Perel JM, Mazariegos GV, Hasegawa SL, Reyes J. Kava-induced fulminant hepatic failure. J Am Acad Child Adolesc Psychiatry 2002;41: 631-2.
6. McKiernan SH, Bavister BD, Tasca RJ. Energy substrate requirements for in-vitro development of hamster 1- and 2-cell embryos to the blastocyst stage. Hum Reprod 1991;6:64-75.
7. Fromenty B, Berson A, Pessayre D. Microvesicular steatosis and steatohepatitis: role of mitochondrial dysfunction and lipid peroxidation. J Hepatol 1997;26: Suppl 1:13-22.
8. Burt AD, Mutton A, Day CP. Diagnosis and interpretation of steatosis and steato-hepatitis. Semin Diagn Pathol 1998;15:246-58.
9. Badizadegan K, Perez-Atayde AR. Focal glycogenosis of the liver in disorders of ureagenesis: its occurrence and diagnostic significance. Hepatology 1997;26: 365-73.
10. Portmann BC, Thompson RJ, Roberts EA, Paterson AC. Genetic and metabolic liver disease. In: Burt AD, Portmann BC, Ferrell LD, eds. MacSween’s pathology of the liver. 5th ed. New York: Churchill Livingstone–Elsevier, 2006:199-326.
11. Boldrini R, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol Res Pract 2004;200:231-40.
12. Mahler H, Pasi A, Kramer JM, et al. Fulminant liver failure in association with the emetic toxin of Bacillus cereus. N Engl J Med 1997;336:1142-8.
13. Luongo MA, Bjornson SS. The liver in ferrous sulfate poisoning; a report of three fatal cases in children and an experimental study. N Engl J Med 1954;251: 995-9.
14. Scriver CR, Beaudet AL, Valle D, et al., eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001.
15. Browning MF, Levy HL, Wilkins-Haug LE, Larson C, Shih VE. Fetal fatty acid oxidation defects and maternal liver disease in pregnancy. Obstet Gynecol 2006;107:115-20.
16. Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet 2006;142C:77-85.
17. Korman SH, Gutman A, Brooks R, Sinnathamby T, Gregersen N, Andresen BS. Homozygosity for a severe novel medium-chain acyl-CoA dehydrogenase (MCAD) mutation IVS3-1G > C that leads to introduction of a premature termination codon by complete missplicing of the
MCAD mRNA and is associated with phenotypic diversity ranging from sudden neonatal death to asymptomatic status. Mol Genet Metab 2004;82:121-9.
18. Yang Z, Lantz PE, Ibdah JA. Post-mor-tem analysis for two prevalent beta-oxidation mutations in sudden infant death. Pediatr Int 2007;49:883-7.
19. Fernandes J, Saudubray J-M, van den Berghe G, Walter JH, eds. Inborn metabolic diseases: diagnosis and treatment. 4th ed. New York: Springer, 2006.
20. Kaminaga T, Mano T, Ono J, Kusuoka H, Nakamura H, Nishimura T. Proton magnetic resonance spectroscopy of Sjögren-Larsson syndrome heterozygotes. Magn Reson Med 2001;45:1112-5.
21. Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Lars-son’s syndrome. AJNR Am J Neuroradiol 1999;20:1671-3.
22. Sijens PE, Westerlaan HE, de Groot JC, et al. MR spectroscopy and diffusion tensor imaging of the brain in Sjögren-Larsson syndrome. Mol Genet Metab 2009;98:367-71.
23. Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain 2001;124:1426-37.
24. Willemsen MA, Van Der Graaf M, Van Der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. AJNR Am J Neuroradiol 2004;25:649-57.
25. Groenendaal F, Bianchi MC, Battini R, et al. Proton magnetic resonance spectroscopy (1H-MRS) of the cerebrum in two young infants with Zellweger syndrome. Neuropediatrics 2001;32:23-7.
26. Bruhn H, Kruse B, Korenke GC, et al. Proton NMR spectroscopy of cerebral metabolic alterations in infantile peroxisomal disorders. J Comput Assist Tomogr 1992;16:335-44.
27. Weller S, Rosewich H, Gärtner J. Cerebral MRI as a valuable diagnostic tool in Zellweger spectrum patients. J Inherit Metab Dis 2008 April 14 (Epub ahead of print).
28. Roomets E, Lundbom N, Pihko H, Heikkinen S, Tyni T. Lipids detected by brain MRS during coma caused by carnitine palmitoyltransferase 1 deficiency. Neurology 2006;67:1516-7.
29. Barker PB, Horská A. Neuroimaging in leukodystrophies. J Child Neurol 2004; 19:559-70.
30. Stone JA, Castillo M. MR in a patient with Zellweger syndrome presenting without cortical or myelination abnor-malities. AJNR Am J Neuroradiol 1998; 19:1378-9.
31. Unay B, Kendirli T, Ataç K, Gül D, Akin R, Gökçay E. Caudothalamic groove cysts in Zellweger syndrome. Clin Dys-morphol 2005;14:165-7.
32. Blau N, Duran M, Blaskovics ME, Gib-son KM, eds. Physician’s guide to the laboratory diagnosis of metabolic diseases. 2nd ed. New York: Springer, 2002.33. Costa C, Costa JM, Slama A, et al. Mutational spectrum and DNA-based prenatal diagnosis in carnitine-acylcarnitine
translocase deficiency. Mol Genet Metab 2003;78:68-73.
34. Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcar-nitine translocase deficiency, clinical, biochemical and genetic aspects. Mol As-pects Med 2004;25:521-32.
35. Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis 2010;33:501-6.
36. American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: toward a uniform screening panel and system — executive summary. Pediatrics 2006;117:S296-S307.
37. Wilson JMG, Jungner G. Principles and practice of screening for disease. Geneva: World Health Organization, 1968.
38. Therrell BL, Johnson A, Williams D. Status of newborn screening programs in the United States. Pediatrics 2006;117: S212-S252.
39. The changing moral focus of newborn screening: an ethical analysis by the President’s Council on Bioethics. Washington, DC: President’s Council on Bioethics, 2008. (http://bioethics.georgetown.edu/pcbe/reports/newborn_screening/ Newborn%20Screening%20for%20the%20web.pdf.)
40. Adamsbaum C, Pinton F, Rolland Y, Chiron C, Dulac O, Kalifa G. Accelerated myelination in early Sturge-Weber syndrome: MRI-SPECT correlations. Pediatr Radiol 1996;26:759-62.
41. Yagishita A, Arai N, Tamagawa K, Oda M. Hemimegalencephaly: signal changes suggesting abnormal myelination on MRI. Neuroradiology 1998;40:734-8.
42. Lamont P, Sachinwalla T, Pamphlett R. Myelin in SIDS: assessment of development and damage using MRI. Pediatrics 1995;95:409-13.