
食管癌发病地域性差异明显,据最新统计我国食管癌发病与死亡病例数均占全球食管癌总发病及死亡病例数的50%以上,是世界平均水平的2倍多[1-2],其中我国河北省磁县、涉县地区食管癌高发。食管鳞状细胞癌患者在我国食管癌患者中占90%以上[3],虽然早期行根治术后预后较好,但多数就诊时已无手术机会,这使我国食管癌患者5年生存率仅为20%左右[4],因此食管鳞状细胞癌的早期诊断及治疗具有重要意义。在长期的临床实践中发现大多数食管癌患者大便干燥,甚至十余日一行,这种肠道环境势必存在菌群的改变。目前已有大量研究证实肠道菌群与结直肠癌、胃癌、肝癌等多种肿瘤密切相关,可作为诊断、治疗及预后的参考评估指标[5-9],但关于食管癌患者肠道菌群特征的研究较少。本研究旨在探索食管鳞状细胞癌患者肠道菌群特征,期望筛选出有统计学差异的特异性肠道菌群,为食管鳞状细胞癌的早期诊断及治疗提供循证依据。
1 对象与方法
1.1 研究对象 选取2022年4—8月就诊于河北医科大学第四医院的食管鳞状细胞癌患者35例作为食管癌组,招募同时期在医院体检的健康志愿者35例作为对照组。本研究已通过河北医科大学第四医院医学伦理委员会审核(伦理审批号:2022KY057)。
1.2 纳入、排除标准 食管癌组纳入标准:(1)经组织病理学诊断为食管鳞状细胞癌,诊断标准参照中国临床肿瘤学会(Chinese Society of Clinical Oncology,CSCO)制订的《中国临床肿瘤学会(CSCO)食管癌诊疗指南-2022》[10];(2)年龄18~75岁;(3)无其他恶性肿瘤病史;(4)未经任何抗肿瘤治疗(如手术、放化疗、免疫治疗等);(5)签署知情同意书。对照组纳入标准:(1)年龄18~75岁;(2)无恶性肿瘤史;(3)签署知情同意书。排除标准:(1)入组前2周内有感染性疾病、抗生素治疗史及益生菌服用史;(2)有精神疾病或无完全民事行为能力;(3)有严重心血管疾病及肠道疾病等(如炎症性肠病、肠易激综合征等)。
1.3 临床资料及标本收集 临床资料收集:收集研究对象的一般资料,包括性别、年龄、饮酒史(经常饮酒:每周饮酒1次以上但达不到每天饮酒;偶尔饮酒:每周饮酒少于等于1次;从不饮酒:从来不饮酒)、吸烟史(经常吸烟:每天吸卷烟1支以上,连续或累计6个月;偶尔吸烟:每周吸卷烟超过4次,但平均每天不足1支;从未吸烟:从来不吸烟)及临床分期(Ⅰ期:T1N0-1M0;Ⅱ期:T2N0-1M0、T3N0M0;Ⅲ期:T3N1M0、T1-3N2M0;Ⅳ期:T4N0-2M0、任何TN3M0、任何T任何NM1)[10]。标本采集:收集研究对象的新鲜粪便标本,嘱受试者将粪便排泄到干净的容器中,避免尿液、马桶壁等对粪便样本的污染,用无菌勺挖取排泄中后部的粪便1 g左右,放入做好标记的冻存管中,立即置于液氮中猝灭约1 min,后将冻存管放入-80 ℃冰箱保存,两组各收集35例受试者粪便标本。
1.4 肠道菌群检测与分析 将收集的70例粪便样本提取基因组DNA,应用16SV34区域特异引物进行聚合酶链式反应(polymerase chain reaction,PCR)扩增(上游引物CCTAYGGGRBGCASCAG,下游引物GGACTACNNGGGTATCTAAT)。将磁珠纯化的PCR产物根据浓度进行等量混样,混匀后使用2%的琼脂糖凝胶电泳检测PCR产物,然后对目的条带用TianGen公司凝胶回收试剂盒(型号:DP214)回收纯化产物。使用Illumina公司建库试剂盒(型号:TruSeq DNA PCR-Free Library Preparation Kit)构建文库,文库定量检查合格后,使用NovaSeq6000进行上机测序。通过对测序序列进行拼接,同时对序列质量进行质控和过滤,以97%的一致性将序列聚类成为分类操作单元(operational taxonomic units,OTUs),然后对OTUs序列与Silva138数据库进行物种注释。根据物种注释情况,进一步分析Alpha多样性与Beta多样性,并进行组间差异的比较,揭示两组群落结构的差异特征。
1.4.1 OTUs分析 基于OTUs注释结果比较两组样本肠道菌群结构的差异。选择两组在门水平和属水平上最大丰度排名前10的物种,绘制物种相对丰度柱形累加图。
1.4.2 物种累积箱形图分析 物种累积箱形图是描述随着样本量的增加物种多样性是否增加的分析,其结果反映持续抽样下新OTUs出现的速率,主要用于判断样本量的选择是否充分合理。
1.4.3 Alpha多样性分析 Alpha多样性主要与丰富度及多样性有关,香农(Shannon)指数和辛普森(Simpson)指数用于描述菌群多样性,Chao1指数和ACE指数用于描述菌群丰富度,Goods_coverage指数描述测序深度。选取以上指数分析两组样本物种分布的多样性和丰富度,并直观展示测序深度。
1.4.4 Beta多样性分析 Beta多样性分析是对两组样本菌群构成情况进行比较分析。主坐标分析(principal co-ordinates analysis,PCoA)是常用的Beta多样性分析方法,是通过一系列的特征值和特征向量排序从多维数据中提取出最主要的元素和结构。基于Unweighted Unifrac距离来进行分析,样本间距离越远代表样本间群落结构差异越大。
1.4.5 线性判别分析及影响因子(linear discriminant analysis effect size,LEfSe)分析 LEfSe分析可展现不同组中丰度有显著差异的物种,用于分析每个物种丰度对差异效果影响的大小。
1.5 统计学方法 采用SPSS 22.0软件对数据进行统计学分析。符合正态分布的计量资料以(x±s)表示,两组间比较采用独立样本t检验;计数资料以相对数表示,两组间比较采用χ2检验。非参数检验Anosim分析用于检验组间差异是否大于组内,判断研究分组是否存在意义。以P<0.05为差异有统计学意义。
2 结果
2.1 基本特征 35例食管鳞状细胞癌患者临床分期为Ⅰ期3例、Ⅱ期6例、Ⅲ期17例、Ⅳ期9例。两组人群性别、年龄、吸烟史、饮酒史比较,差异均无统计学意义(P>0.05),见表1。
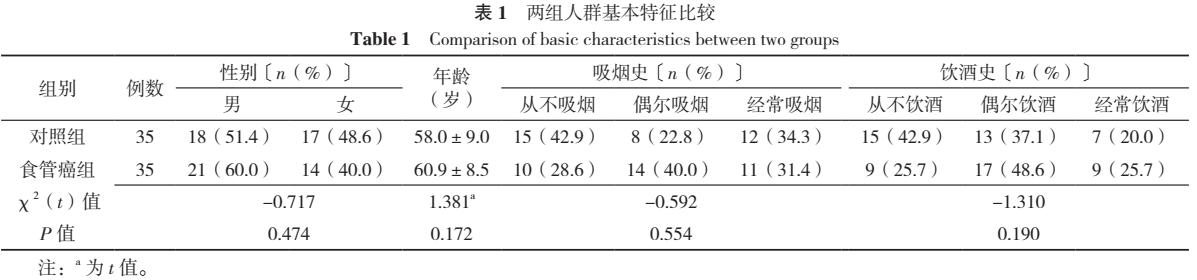
2.2 肠道菌群OUTs分析 对获得的有效数据进行分析共得到3 602个OTUs,再对OTUs序列进行物种注释,共有52个OTUs注释到物种门水平(Phylum),756个OTUs注释到物种属水平(Genus)。门水平上两组人群以厚壁菌门(Firmicutes)、放线菌门(Actinobacteriota)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidota)、疣微菌门(Verrucomicrobiota)为主;与对照组相比,食管癌组Bacteroidota及Actinobacteriota丰度比例升高,而Firmicutes、Verrucomicrobiota丰度比例明显降低(图1A)。属水平上两组人群以双歧杆菌属(Bifidobacterium)、大肠杆菌-志贺菌属(Escherichia-Shigella)、栖粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、普雷沃菌属(Prevotella)、克雷伯菌属(Klebsiella)、链球菌属(Streptococcus)、经黏液真杆菌属(Blautia)、艾克曼菌属(Akkermansia)、戴阿利斯特杆菌属(Dialister)为主;与对照组相比,食管癌组Faecalibacterium丰度比例明显降低(图1B)。
2.3 肠道菌群物种累积箱形图分析 物种累积箱形图位置趋于平缓,两组样本量充分,测序数据量合理,随着样本量的增加肠道菌群物种并不会显著增多,见图2。
2.4 肠道菌群Alpha多样性分析 两组Shannon、Simpson、Chao1、ACE、Goods_coverage指数比较,差异均无统计学意义(P>0.05),见表2;Goods_coverage指数两组测序深度均达99%以上,数据量充足。
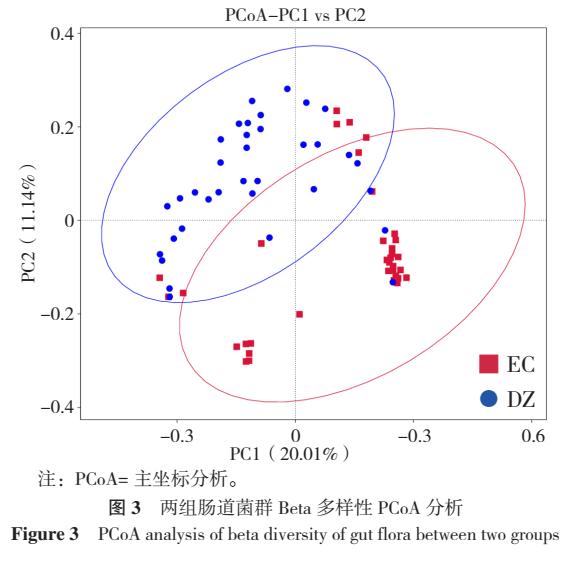
2.5 肠道菌群Beta多样性分析 PCoA图结果显示,食管癌组与对照组样本整体上相距较远,两组样本间群落结构差异较大,见图3。食管癌组与对照组肠道菌群群落结构比较,差异有统计学意义(t=10.837,P<0.001)。
2.6 肠道菌群物种差异分析
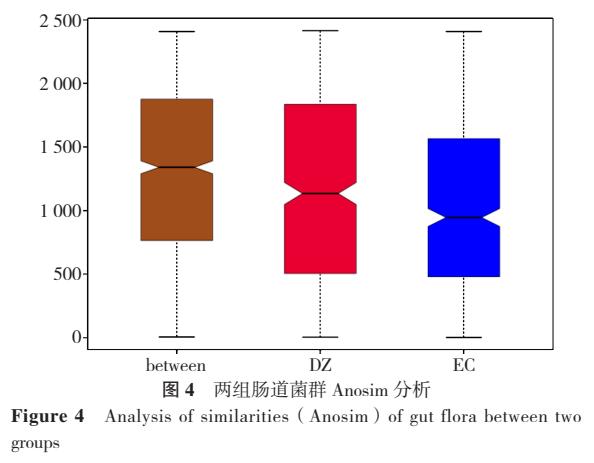
2.6.1 组间与组内差异性分析 两组肠道菌群物种差异组间大于组内,差异有统计学意义(R=0.158,P<0.05),见图4。
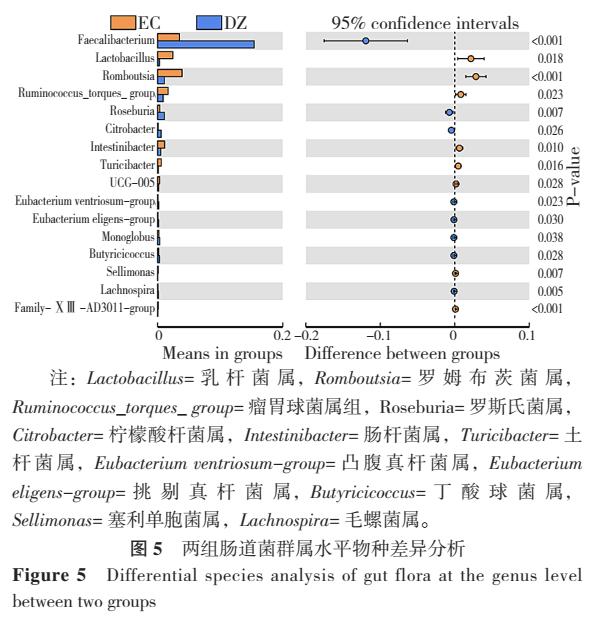
2.6.2 属水平肠道菌群物种差异分析 在属水平上,两组肠道菌群中16类菌属的丰度比较,差异有统计学意义(P<0.05)。两组肠道菌群相对丰度较高的前8位菌属中,食管癌组Faecalibacterium、罗斯氏菌属(Roseburia)、柠檬酸杆菌属(Citrobacter)的丰度低于对照组,乳杆菌属(Lactobacillus)、罗姆布茨菌属(Romboutsia)、瘤胃球菌属组(Ruminococcus_torques_group)、肠杆菌属(Intestinibacter)、土杆菌属(Turicibacter)的丰度高于对照组(P<0.05),见图5。食管癌组中Lactobacillus丰度高于对照组,经询问采样前2周时补充过肠道益生菌者要多于对照组(食管癌组11例,对照组6例),所以本研究将乳酸杆菌作为干扰因素,不纳入组间物种差异分析。
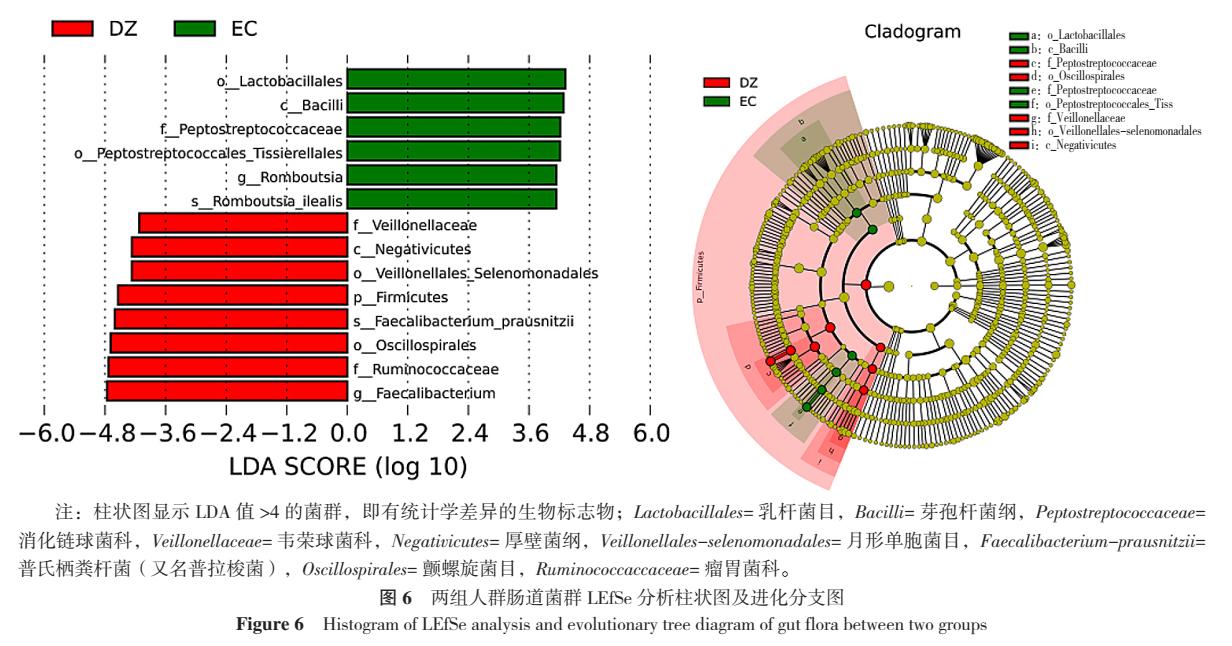
2.6.3 肠道菌群LEfSe分析 柱状图及进化分支图结果显示,两组肠道菌群中14种肠菌的丰度比较,差异有统计学意义(P<0.05)。其中食管癌组乳杆菌目(Lactobacillales)、芽孢杆菌纲(Bacilli)、消化链球菌科(Peptostreptococcaceae)、Peptostreptococcales-Tissierellales、Romboutsia、Rombutsia-ilealis丰度高于对照组(P<0.05);对照组韦荣球菌科(Veillonellaceae)、厚壁菌纲(Negativicutes)、月形单胞菌目(Veillonellales-selenomonadales)、Firmicutes、普氏栖粪杆菌(又名普拉梭菌Faecalibacterium-prausnitzii)、颤螺旋菌目(Oscillospirales)、瘤胃菌科(Ruminococcaccaceae)、Faeculibacterium丰度高于食管癌组(P<0.05)。在属及种水平上与对照组相比,食管癌组中Faeculibacterium及Faecalibacterium-prausnitzii丰度降低,Romboutsia及Rombutsia-ilealis丰度升高,是具有重要鉴别意义的生物标志物(P<0.05),见图6。
3 讨论
正常人体肠道内约有100万亿个肠菌,总重为1~2 kg,被称为最大共生体[11]。这些共生肠菌在人体内发挥了重要作用,如消化吸收、免疫调控、维持肠道黏膜屏障及肠道稳态等[12]。已知正常人体内肠菌在门水平上Firmicutes和Bacteroidota约占90%,Actinobacteriota、Proteobacteria和梭杆菌门(Fusobacteriota)等约占10%[13]。既往大量研究发现肠道菌群通过多种方式影响多种肿瘤的发生和发展[5-9]。
本研究结果显示,食管癌组与对照组人群肠道菌群Alpha多样性即菌群物种丰富度未见明显差异,而Beta多样性即两组菌群群落结构存在明显差异。两组肠道菌群以Firmicutes、Actinobacteriota、Proteobacteria、Bacteroidota、Verrucomicrobiota为主。与对照组相比,食管癌组Bacteroidota及Actinobacteriota丰度比例升高,而Firmicutes、Verrucomicrobiota比例明显降低。两组人群肠道菌群T-test检验结果在属水平共有16类菌属的丰度存在明显差异,相对丰度较高的前8位菌属中,食管癌组Faecalibacterium、Roseburia、Citrobacter丰度低于对照组;Lactobacillus、Romboutsia、Ruminococcus_torques_group、Intestinibacter、Turicibacter丰度明显高于对照组。LEfSe分析结果显示14种肠菌丰度存在差异,在属及种水平上与对照组相比,食管癌组Faeculibacterium及Faecalibacterium-prausnitzii丰度明显降低,Romboutsia及Rombutsia-ilealis丰度明显升高。
Faeculibacterium是本研究中表达丰度较高且变化最明显的菌属,可能是对食管癌发生具有重要意义的差异菌属。有研究发现Faeculibacterium在乳腺癌患者中表达丰度明显降低,与代谢物磷胆碱结合,可能成为乳腺癌检测的新方法[14]。另外发表在Science上的一项研究也发现,Faeculibacterium在PD-L1治疗无效的黑色素瘤患者中丰度较低,可能与上调CD8+ T淋巴细胞等免疫细胞和抗原呈递相关分子促进抗PD-1肿瘤治疗有关[15]。Faeculibacterium中最著名的Faecalibacterium-prausnitzii占正常人体粪便菌群总数的5%~15%,是人体最重要的肠菌之一,也是丁酸的重要生产者之一[16-17]。
有研究发现,与健康人群相比,在结肠癌患者粪便中Faecalibacterium-prausnitzii含量显著降低,该研究还推测可能与其产生的丁酸盐减少有关[18]。同时体外研究发现Faecalibacterium-prausnitzii及其培养上清液具有抑制结肠癌细胞增殖及促进凋亡等作用[19]。Faecalibacterium-prausnitzii还能够抑制乳腺癌细胞中白介素6的分泌和Janus激酶2/信号转导和转录激活因子3的磷酸化,从而抑制乳腺癌增殖和侵袭,促进其凋亡[14]。本研究Faeculibacterium及Faecalibacterium-prausnitzii的变化均与上述研究一致。
ZHANG等[20]及SEOL等[21]研究发现Romboutsia在结直肠癌及胃癌患者中丰度明显升高,本研究结果与之一致。其分类中的Romboutsia-ilealis被认为是葡萄糖代谢的潜在恶化剂,是2型糖尿病的危险因素[22],但与肿瘤的关系研究很少。WANG等[23]研究发现,Roseburia在结肠癌患者中大量减少,可能通过其产生的丁酸盐阻断核因子κB通路的活化。ZHENG等[24]发现瘤胃球菌属(Ruminococcus)在肺癌患者肠道中含量较高。肿瘤坏死因子可以直接*伤杀**和抑制肿瘤细胞;IIDA等[25]研究发现Ruminococcus与肿瘤坏死因子分泌呈正相关,以上研究结果均表明肠道菌群的改变与肿瘤的发生、发展及治疗密切相关。其中Ruminococcus的变化趋势与本题组之前研究的食管癌原位模型小鼠的肠道菌群改变相同[26],可以相互验证。但其中部分差异菌属不同,因为人类肠道菌群受饮食、疾病、药物等外界影响因素较多,如以往研究表明乳酸杆菌属于益生菌[27],而本次研究中乳酸杆菌在食管癌患者中要高于对照组人群,这与以往研究结果不太一致,通过询问受试者生活饮食,虽近2周未服用益生菌,但有11例食管癌患者有长期服用乳酸菌奶制品史,数量多于对照组,提示需要进一步研究证实,并在今后研究肠道菌群时要延长停用益生菌的时间。
综上,食管鳞状细胞癌患者与正常人群之间存在显著性差异肠菌,其中部分特异性改变菌群可能与食管癌发生密切相关,并可能为食管癌的诊断及治疗提供参考依据。本研究局限性:首先,食管鳞状细胞癌患者均为本院就诊患者,地域性单一,样本量偏小,可能结果需要多地区大样本的临床研究验证;其次,本研究应用的16S rDNA测序主要研究群落的多样性、物种构成及其之间的进化关系,但该测序分析不是宏基因组测序,所以不能进行基因和功能等层面的深入研究;再次,目前研究只是揭示了食管鳞状细胞癌患者与正常人群肠道菌群的变化,但并未从机制上进行深入的研究验证;最后,对于标本采样前抗生素、益生菌的停用时间目前无统一标准,但根据本研究结果建议应适度延长,具体时限有待进一步研究确认。
本文无利益冲突。
参考文献略
本文来源:张玉双,孔令洋,管佳畅,等. 基于16S rDNA测序探索食管鳞状细胞癌患者肠道菌群特征[J]. 中国全科医学,2023,26(20):2496-2502. DOI:10.12114/j.issn.1007-9572.2022.0832.