在阅读此文前,诚邀您请点点右上方的“关注”,既方便您进行讨论与分享,还能及时阅读最新内容,感谢您的支持。
胃肠道微生物是维持哺乳动物机体内环境平衡的重要因素,在维持宿主各项生命活动与生理健康方面发挥着不可替代的作用。
哺乳动物的胃肠道中寄宿着超过1 000种的复杂的微生物群,其数量可达1014,约为动物体细胞总数的10倍。
大量研究显示胃肠道微生物能够参与宿主的机体免疫、营养吸收和代谢调控等生理活动,肠道微生物区系紊乱与炎性肠病、肥胖、糖尿病、结直肠癌等一系列疾病密切相关。
因此,加强对胃肠道微生物的研究、了解其组成,对于进一步阐明胃肠道微生物群落变化与疾病的相互关系、推进疾病的临床诊断与治疗具有重要意义。

高通量测序技术的快速发展和广泛应用,为系统研究动物胃肠道微生物的组成、结构和功能提供了极大助力。
目前,利用宏基因组测序技术,多种哺乳动物的胃肠道微生物区系被研究和了解,包括人、小鼠、山羊、牛、猪等。
尽管对动物胃肠道微生物菌群的研究越来越多,但这些研究中所使用的样本多来自于粪便,其研究结果受宿主遗传、肠道内外部环境等因素的影响。
因此,目前对哺乳动物各段胃肠道微生物菌群的了解仍然有限。
Sprague-Dawley大鼠又称SD大鼠,在1925年由美国Sprague-Dawley农场选用Wistar大鼠培育而成。

作为最常用的实验动物之一,Sprague-Dawley大鼠被广泛应用于营养代谢、肿瘤、内分泌等方面的研究。
因此,充分了解Sprague-Dawley大鼠的胃肠道微生物菌群的组成和分布,是进行相关试验研究的重要前提。
然而,目前对Sprague-Dawley大鼠胃肠道微生物群落组成的研究相对较少,仍需对其作进一步探索和补充。
本研究利用宏基因组测序技术,通过分析6只雄性Sprague-Dawley大鼠各肠段内容物中的微生物群落组成,了解Sprague-Dawley大鼠胃肠道微生物的种类、分布结构和丰富度的差异,以期能为以Sprague-Dawley大鼠为动物模型的相关研究提供微生物群落组成的参考。
材料与方法
试验动物
6只4周龄SPF级 Sprague-Dawley雄性大鼠,体重均为110 g左右,购自北京维通利华实验动物技术有限公司。
主要试剂及仪器
E.Z.N.A. DNA提取试剂盒,Axy Prep DNA提取试剂盒。
Nano Drop 2000紫外可见分光光度计,热循环PCR系统,Quanti FluorTM-ST荧光计,Illumina MiSeq测序平台。

大鼠胃肠道样品采集
6只4周龄SPF级Sprague-Dawley雄性大鼠,按照SPF级标准于徐州医科大学实验动物中心喂养28 d。
大鼠禁食10 h后进行脱颈椎处死,在无菌条件下分别取大鼠胃、十二指肠、空肠、回肠、盲肠、结肠和直肠的肠道内容物置于2 mL无菌EP管中,称取每只大鼠同一肠段的等重(20 mg)肠腔内容物,加入500 μL灭菌水充分混合,于-80 ℃储存。
样本DNA提取与PCR扩增
使用E.Z.N.A. DNA提取试剂盒从大鼠肠道内容物样本中提取微生物DNA。
使用Nano Drop 2000紫外可见分光光度计纯化和测定DNA浓度, 1%琼脂糖凝胶电泳检测DNA质量。
利用热循环PCR系统对细菌16S rRNA基因的V3~V4高变区分别用338F(5′-ACTCCTAGGGGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)[17]引物进行扩增。
PCR反应程序如下:95 ℃预变性3 min; 95 ℃变性30 s, 55 ℃退火30 s, 72 ℃延伸45 s, 共27个循环;72 ℃最终延伸10 min。
PCR反应在20 μL体系中进行,其中含有5× FastPfu缓冲液4 μL, 2.5 mmol/L dNTPs 2 μL,5 μmol/L每种引物0.8 μL,0.4 μL Fast Pfu聚合酶以及10 ng模板DNA。
使用2%琼脂糖凝胶中提取PCR产物,使用Axy Prep DNA提取试剂盒进一步纯化,并用Quanti FluorTM-ST对DNA进行量化。
测序数据处理
原始fastq文件由Trimmomatic进行质量过滤,并由FLASH按照以下标准进行合并:(1)在50 bp滑动窗口中,任何接收到平均质量分数小于20的站点的读数都被截断;(2)重叠长度大于10 bp的序列按重叠合并,错配不大于2 bp; (3)根据文库(精确匹配)和引物(允许2个核苷酸不匹配)分离每个样本的序列,并去除含有不明确碱基的读码。
使用UPARSE(http: //drive5.com/UPARSE/)对操作分类单元(OTU)进行聚类,相似度截止为97%。
用RDP分类器算法(http: //RDP.cme msu.edu/)根据SILVA数据库对每个16S rRNA基因序列进行分类分析, 置信阈值70%。
结果与分析
α多样性分析
为研究大鼠肠道微生物群落的丰度和多样性,对肠道各部位内容物进行α多样性分析,各指数计算结果见表1。
每个样本的覆盖率(coverage)均大于或等于0.99(表1),表明所测序的样本量具有代表性。
十二指肠内的微生物群落最少,多样性最低;盲肠、结肠和直肠样本的细菌群落丰富度和多样性指数较为相似,且指数值高于肠道其它部位;十二指肠、空肠和回肠样本细菌群落的丰富度和多样性指数差异较大,指数值低于胃和大肠部位;大肠部位的整体的微生物群落的丰富度和多样性比小肠部位更稳定。
表1 大鼠胃肠道不同区域微生物的α多样性分析
|
区域 Region |
Sobs |
Shannon |
Simpson |
Ace |
Chao |
Coverage |
|
胃Stomach |
537 |
2.95 |
0.13 |
621.95 |
615.01 |
0.996 |
|
十二脂肠Duodenum |
48 |
0.80 |
0.70 |
205.97 |
103.20 |
0.999 |
|
空肠Jejunum |
463 |
2.70 |
0.33 |
537.50 |
531.25 |
0.997 |
|
回肠Ileum |
318 |
1.49 |
0.57 |
391.19 |
388.66 |
0.997 |
|
盲肠Cecum |
683 |
4.78 |
0.02 |
749.81 |
746.76 |
0.996 |
|
结肠Colon |
779 |
4.30 |
0.08 |
799.75 |
799.41 |
0.998 |
|
直肠Rectum |
699 |
3.70 |
0.09 |
808.31 |
819.48 |
0.994 |
大鼠胃肠道内容物群落组成分析
用RDP分类器(贝叶斯算法)对具有97%相似性的代表性OTU序列进行分类分析,确定了29个菌门。
分析结果显示,29个菌门在胃肠道不同部位的分布存在差异,在胃、十二指肠、空肠、回肠、盲肠、结肠和直肠中分别分布20、5、10、8、17、23和16个菌门。
通过对不同菌门的含量进行分析发现,在门水平,大鼠胃肠道菌群主要由厚壁菌门(49.44~99.89%)组成。
其中,厚壁菌门在十二指肠中最丰富(99.89%),在盲肠中最少(45.41%);拟杆菌门在盲肠中最丰富(39.53%),在十二指肠中最少(0.01%);变形菌门在直肠中最丰富(33.26%),在十二指肠中最少(0.02%);放线菌门在结肠中最丰富(9.53%),在十二指肠中最少(0.08%);其它菌门的相对丰度较低。
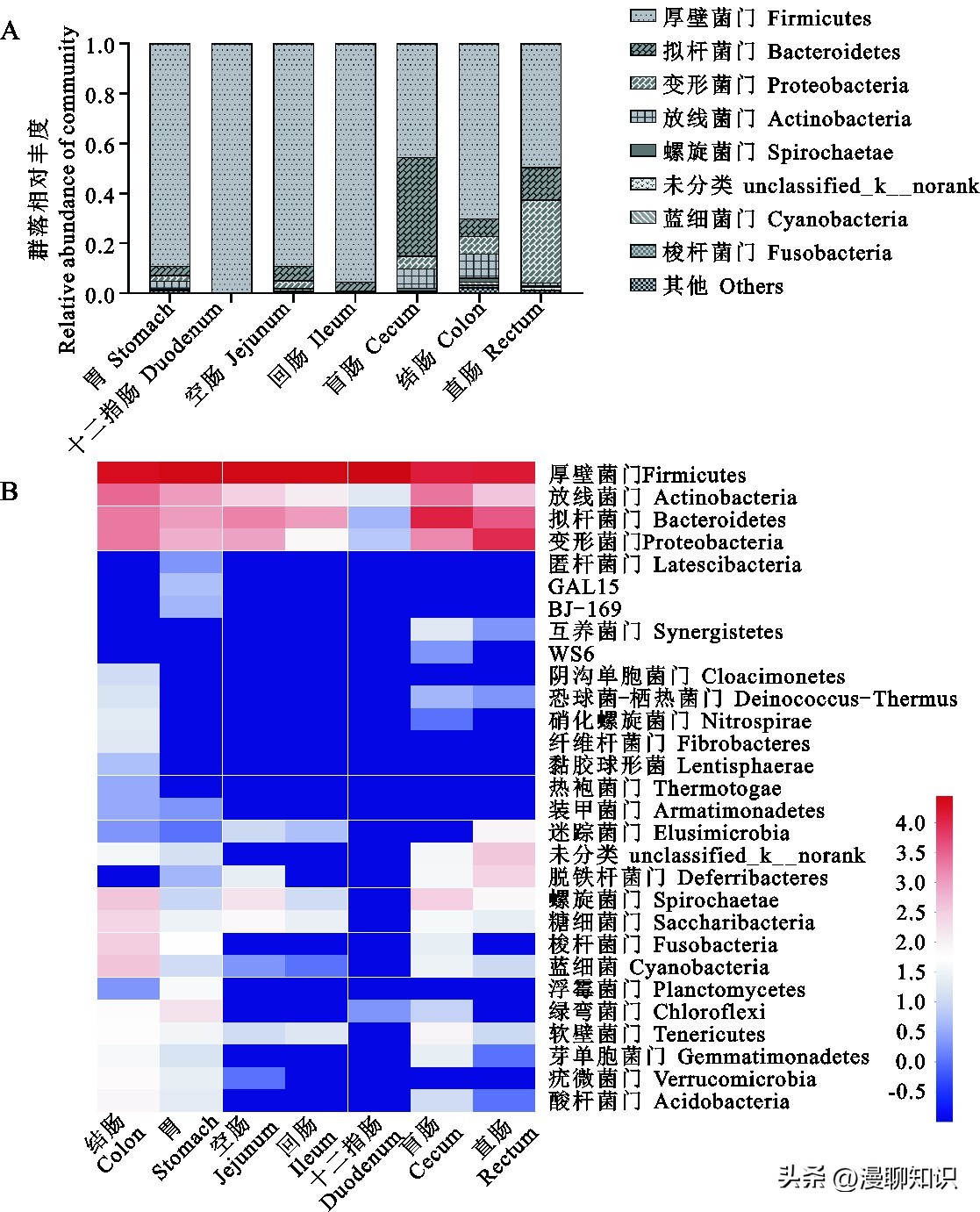
图1 门水平大鼠胃肠道不同部位的细菌组成
A.柱状图(相对丰度小于0.01的细菌分类群均归为“其他”类);B. heatmap图(细菌分类单元数的lg值用颜色强度表示)
为了进一步研究大鼠胃肠道细菌的组成,在属水平上分析了细菌群落的组成和变化,并通过序列比对鉴定了459个菌属。
分析结果显示,乳杆菌属Lactobacillus是胃肠道各部位的优势菌属,其丰富度在各肠道有所差异(。
乳杆菌属在十二指肠中最丰富(96.95%),在盲肠中最少(17.70%)。
拟杆菌属Bacteroides在盲肠中最丰富(14.66%),螺杆菌属Helicobacter在直肠中最丰富(13.25%)。

在大鼠胃中,>1%的属有乳酸杆菌(67.68%)、未分类的消化链球菌科(6.60%)、链球菌属(2.07%)和罗氏菌属Rothia(1.19%);十二指肠中,>1%的属有乳杆菌属(96.95%)、未分类的消化链球菌科(2.20%);空肠中,>1%的属为乳酸杆菌属(62.40%)、未分类的消化链球菌科(4.86%)、未分类的毛螺菌科(4.22%)、(2.72%)、奎因氏菌属(2.50%)、螺杆菌属(1.88%)、毛螺菌NK4A136群(1.80%)、拟杆菌属(1.74%)、瘤胃球菌(1.68%)和脱硫弧菌属Desulfovibrio(1.03%);回肠中,>1%的属有乳杆菌属(82.23%)、未分类的消化链球菌科(5.08%)、_S24-7_group(2.56%)和瘤球菌UCG-005(1.88%)。
在盲肠中>1%的属包括乳杆菌属(17.70%)、拟杆菌属(14.66%)、理研菌科RC-9属(13.25%)、链球菌属(5.26%)、未分类的拟杆菌目(4.51%)、罗氏菌属(4.05%)、未分类的消化链球菌科(2.77%)、(1.94%)、格鲁比卡氏菌属(1.42%)、梭菌属(1.29%)、未分类的毛螺菌科(1.19%)。
在结肠中>1%的属包括乳杆菌属(35.41%)、链球菌属(6.22%)、未分类的消化链球菌科(5.35%)、未分类的毛螺菌科(3.96%)、大肠杆菌-志贺氏菌属(3.14%)、双歧杆菌属(3.00%)、(1.82%)、普雷沃菌-9 (1.72%)、肠杆菌属(1.41%)、拟杆菌属(1.32%)、罗斯伯里氏菌属(1.04%)、格鲁比卡氏菌属(1.04%)、罗斯氏菌属(1.01%)、科林斯式菌属(1.00%)。
最后,在直肠中>1%的属是螺杆菌属(30.75%)、乳杆菌属(29.75%)、(4.13%)、未分类的消化链球菌科(3.43%)、拟杆菌属(3.13%)、瘤球菌UCG-005(2.80%)、脱硫弧菌属(1.31%)、普雷沃菌-9属(1.54%)、理研菌科RC-9属(1.31%)、克里斯滕森菌R-7群(1.08%)。
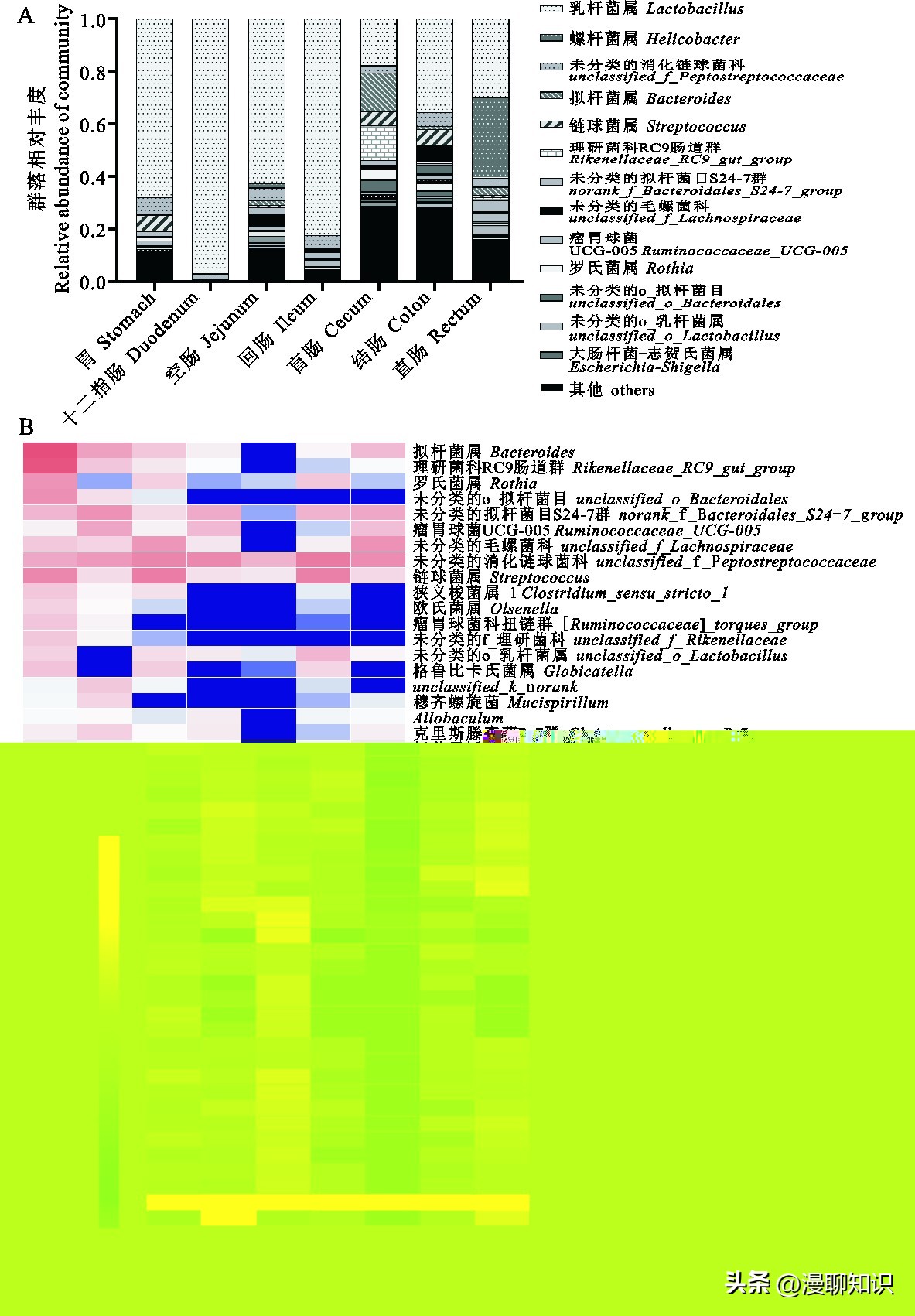
图2 属水平大鼠胃肠道不同部位的细菌组成
A.柱状图(相对丰度小于0.01的细菌分类群均归为“其他”类);B. heatmap图(细菌分类单元数的lg值用颜色强度表示)
β多样性分析
PCoA分析结果显示,十二指肠、空肠和回肠内的菌群有聚集的趋势,小肠内的细菌组成相似,胃和结肠的细菌组成相似。
盲肠、直肠内的细菌群落组成结构与其它部位菌群相比差异较大。
测序样品的稀释曲线已进入平台期,达到饱和状态,表明本试验的测序数据量足够充分,可以反映样品中绝大多数微生物物种信息。
从聚类分析结果来看,十二指肠、空肠及回肠样品聚集成1簇,盲肠和直肠各部位聚集成1簇,胃和结肠样品聚集成1簇,说明盲肠和直肠的细菌群落组成相似性较高,十二指肠内容物中的细菌群落组成与回肠、空肠相似性较高,小肠与大肠的微生物菌落组成差异较大。
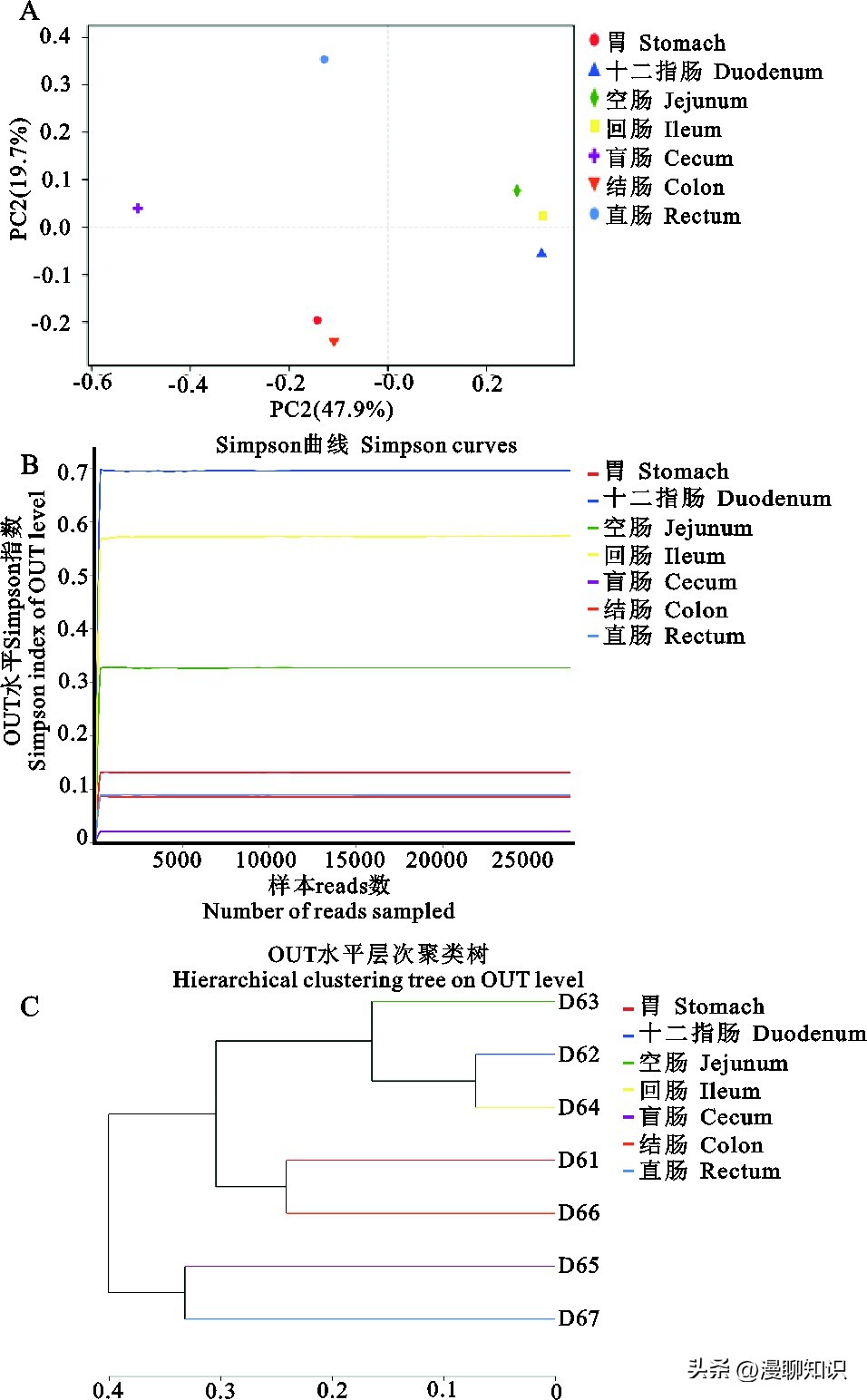
图3 胃肠道不同部位细菌组成及相互关系的差异
A.主坐标分析(PCoA);B. Simpson稀释曲线图;C.样本层次聚类分析树状图
肠道微生物是维持哺乳动物机体内环境平衡的重要因素,在维持宿主各项生命活动与生理健康方面发挥着不可替代的作用。
Sprague-Dawley大鼠作为常用的实验动物,经过长期的选择性繁殖,相对于其他实验动物具有遗传背景稳定、易于操作等优点。
然而,关于Sprague-Dawley大鼠肠道微生物相关描述的研究仍相对有限。
因此,本研究采用宏基因组测序技术对Sprague-Dawley大鼠胃肠道微生物的组成和分布进行分析。

研究结果表明,Sprague-Dawley大鼠肠道微生物的种类超过450种,其中厚壁菌、拟杆菌、蛋白质菌和放线菌是Sprague-Dawley大鼠胃肠道中的优势菌门,乳酸杆菌是胃肠道中的优势菌属,结肠内的微生物最为丰富。
其中厚壁菌主要聚集在小肠中,放线菌、拟杆菌和蛋白质菌主要聚集在大肠部位,这与前人的研究结果具有一致性。
细菌分布的聚集性现象可能是由于消化道不同部位的内环境差异所造成的。
上消化道与下消化道在pH、氧浓度及代谢物质方面的差异导致了乳杆菌属在小肠内大量聚集,而拟杆菌属、链球菌属及螺杆菌属在大肠内聚积。
在本研究中螺杆菌属主要聚集于直肠部位而非盲肠,盲肠中的拟杆菌属和理研菌科RC-9属含量明显高于其他部位,这有别于Li等[16]的研究结果。
产生不同结果的原因可能是饲料和动物所处环境的不同,这就提示我们在实际的应用中即使是同种动物的胃肠道微生物菌群也不是一成不变的,其受到各种环境因素的影响。
大鼠大肠部位的微生物的物种丰富度和多样性普遍高于小肠,大鼠结肠内微生物的物种丰富度最高,十二指肠内的微生物物种丰富度最低,这与Gu等和Han等的研究结果有所差异,后者认为十二指肠样本中的菌门最丰富,并推测十二指肠中“暂时性微生物群”的存在和“消失”可以解释其较高的多样性。
事实上,除少量耐酸性细菌外,大部分微生物在进入十二指肠之前会在胃的酸性条件下死亡,因此进入十二指肠的微生物相对有限。
除此之外,十二指肠内的液体组成复杂,包括胆汁、胰外分泌液等成份,其含有的大量的蛋白酶不利于肠道微生物的稳定增殖,这就解释了十二指肠微生物物种丰富度最低的原因。
肠蠕动使绝大多数细菌被送入下段肠道,结肠内的消化酶含量相对较低,利于厌氧细菌的增殖,因而结肠内的微生物最为丰富。
Gu等的研究认为,环境中的微生物可以通过饮食的方式聚积于动物胃中,这些微生物在胃的酸性条件下死亡后,其DNA可以暂时保持不变并转移到十二指肠中,干扰了十二指肠原本的细菌组成,使用基于DNA的方法进行检测可能导致许多细菌的检测结果出现假阳性。

胃肠道不同部位生理功能与环境的差别,决定了小肠和大肠之间细菌群落的差异。
大肠主要负责水的吸收,而小肠则主要负责营养物质的吸收,二者在结构与生理功能上的差异主要表现在PH值、营养物质、氧浓度、肠蠕动、物质分泌与代谢等多方面。
因此,小肠内环境比大肠更为复杂多变,这可能是小肠各肠段菌群在组成和结构上的差异性高于大肠的原因,大肠的功能相对单一决定了大肠内微生物丰富度和多样性较为稳定。
并且相比大肠环境而言,小肠内粘液的pH值更接近胃酸、氧浓度更高,这也解释了Sprague-Dawley大鼠小肠内兼性厌氧菌含量更高,乳酸杆菌在小肠内的丰富度明显高于大肠的结果,这与之前的研究保持一致。

除此之外,我们还发现Sprague-Dawley大鼠的胃肠道细菌结构有别于小鼠等动物。
这些结果表明,宿主的遗传可以影响胃肠道微生物种群。
Zoetendal等比较了不同个体之间的肠道细菌结构,发现具有相同遗传背景的个体之间的细菌群落高度相似。
Jussi等对两种小鼠肠道菌群结构进行分析,发现BALB/c小鼠与C57BL/6J小鼠肠道菌群存在显著差异。
Ley等还比较了不同基因型小鼠肠道细菌结构,证明遗传改变会影响细菌群落的多样性。
总之,Sprague-Dawley大鼠胃肠道中含有种类丰富、数量巨大的微生物,这些微生物的组成及分布受胃肠道内的位置影响,这种影响是由肠道各部位的功能及环境所决定的。
了解Sprague-Dawley大鼠胃肠道菌群的组成,对于指导以大鼠为实验动物模型的研究具有重要意义。
本研究通过宏基因组测序技术,比较了Sprague-Dawley大鼠胃肠道各肠段微生物群落的组成及分布。
研究结果发现大鼠胃肠道各肠段的细菌群落不同。
乳酸杆菌是Sprague-Dawley大鼠各胃肠道的优势菌属。
大鼠盲肠微生物菌群的丰富度最高,十二指肠微生物菌群的丰富度最低,大肠微生物的菌群丰富度和多样性高于小肠,且微生物菌群组成比小肠更稳定。
本试验的研究结果阐述了Sprague-Dawley大鼠胃肠道不同区域的细菌组成和分布,并与前人的试验结果进行对比,有助于提高我们对这一广泛应用的实验动物的微生物生态的理解。
[1] HEINTZ-BUSCHART A,WILMES P.Human gut microbiome:Function matters[J].Trends Microbiol,2018,26(7):563-574.
[2] SENDER R,FUCHS S,MILO R.Revised estimates for the number of human and bacteria cells in the body[J].PLoS Biol,2016,14(8):e1002533.
[3] LYNCH S V,PEDERSEN O.The human intestinal microbiome in health and disease[J].N Engl J Med,2016,375(24):2369-2379.
[4] THAISS CA,ZMORA N,LEVY M,et al.The microbiome and innate immunity[J].Nature,2016,535(7610):65-74.